Поиск полиморфизмов.
1. Анализ качества чтений и их очистка
По заданию мне досталась 17 хромосома человека chr17.fasta и файл с ридами для
этой хромосомы chr17.fastq (без адаптеров).
С помощью программы Trimmomatic
была произведена очистка чтений:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr17.fastq chr17_out.fastq TRAILING:20 MINLEN:50
Таким образом, в выходном файле chr17_out.fastq с конца каждого чтения отрезаны
нуклеотиды с качеством ниже 20 (TRAILING:20), а также оставлены только чтения длиной не
меньше 50 нуклеотидов (MINLEN:50).
С помощью программы
FastQC я сравнила качество изначального чтения и полученного после очистки:
fastqc *.fastq
где вместо * был вписан либо исходный файл chr17.fastq, либо файл с ридами после
чистки chr17_out.fastq. В итоге было получено два архива, а также 2 .html файла
с отчетами: chr17_fastqc.html и
chr17_out_fastqc.html. На рис. 1-4 представлены
основные характеристики для полученных файлов с чтениям.
Прим: сначала я
просмотрела характеристику чтений до очистки, чтобы понять, нужна ли очистка, но
обе характеристики приведены уже после очистки рядом друг с другом для удобства
☺
 Рис. 1. Общая статистика файла с ридами
до чистки Рис. 1. Общая статистика файла с ридами
до чистки |
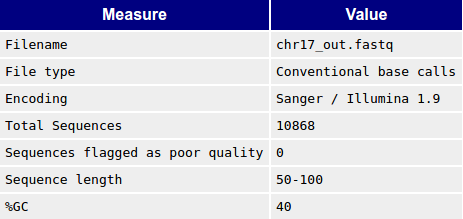 Рис. 2. Общая статистика файла с ридами
после чистки Рис. 2. Общая статистика файла с ридами
после чистки |
|
|
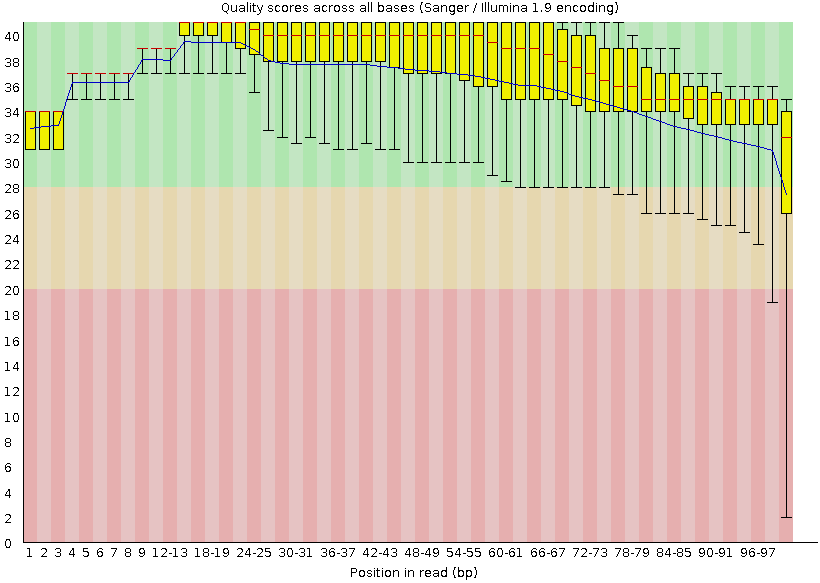 Рис. 3. Качество отдельных
п.н. в ридах до чистки Рис. 3. Качество отдельных
п.н. в ридах до чистки |
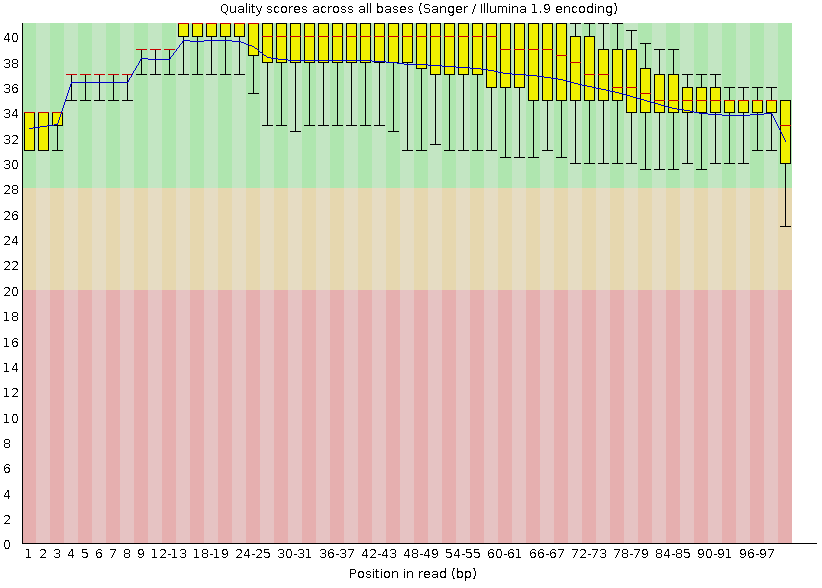 Рис. 4. Качество отдельных
п.н. в ридах после чистки Рис. 4. Качество отдельных
п.н. в ридах после чистки |
| Как видно из рис. 3 и 4 после чистки, показатели качества п.н. в ридах
стали намного лучше. Таким образом, мы получили только те чтения, качество которых нас
устраивает, и с которыми можно работать дальше. |
Помимо приведенных изображений в отчете содержатся и другие (случаи, когда все хорошо, обозначены
✔; когда сомнительно (warning) — !; когда все плохо (failure)
— ✖):
- ✔Per sequence quality scores — распределение среднего качества
среди ридов. В нашем случае все хорошо: пик на 38, т.е. качество у большинства ридов
достаточно хорошее.
- ! Per base sequence quality — процент каждого нуклеотида в
ридах. В рандомной библиотеке процент разных нуклеотидов для всех позиций, по идее, должен
быть примерно одинаковым, так что линии на графике должны быть параллельными. Если каких-то
нуклеотидов в последовательности больше, то это отразится в более высоко расположенной
кривой, но параллельность все равно должна сохраняться. В нашем случае неровности
относительно сильны, так что, возможно, это должно заставить задуматься о причинах.
- ✖Per sequence GC content — распределение GC-состава для всех
ридов. Для нормальной рандомной библиотеки распределение GC-состава должно быть нормальным,
т.е. центральный пик должен совпадать с пиком для GC-состава всего генома. Т.к. исходного
генома нет, вместо него используется геном, построенный с помощью расчетов из имеющихся
данных. В нашем случае этот модуль отмечен как failure, т.к. отклонение
от нормального (гауссовского) распределения составляет более 30% ридов. Причиной может быть
загрязненность библиотеки.
- ✔Per base N content — график, отражающий позиции, в которых
был неопределен нуклеотид (N). В нашем случае все хорошо (для всех позиций 0 (< 5%)).
- ! Sequence Length Distribution — график, отображающий распределение
длин ридов. В нашем случае модуль обозначен warning. Должен быть один пик для
какой-то длины, а в нашем случае в библиотеке соотносимое число ридов с различающимися
длинами (т.е. есть несколько вариантов преобладающих длин), что странно, т.к. обычно
используются риды одной длины.
- ✔Sequence Duplication Levels
- ✔Overrepresented sequences
- ✔Adapter Content
- ✔Kmer Content
2. Картирование чтений. Анализ выравнивания
Для картирования чтений использовалась программа
BWA. Сначала я проиндексировала референсную последовательность chr17.fasta:
bwa index chr17.fasta
Затем было построено выравнивание прочтений chr17_out.fastq и проиндексированного
референса в формате .sam с использованием алгоритма mem:
bwa mem chr17.fasta chr17_out.fastq > align.sam
После этого с помощью пакета samtools файл с выравниванием был переведен в бинарный
формат .bam (сначала просто команда без параметров, чтобы посмотреть описание):
samtools view align.sam -b -o align.bam
Получившийся файл align,bam был подан на вход другой команде, чтобы отсортировать
выравнивание чтений с референсом по координате в референсе начала чтения:
samtools sort align.bam -T wtf.txt -o al_sort.bam
Параметр -T был задан для записи временных файлов в файл wtf.txt, т.к. иначе они
выдавались в stdout. Далее полученный файл был проиндексирован:
samtools index al_sort.bam
А затем я выяснила, сколько чтений откартировалось на геном:
samtools idxstats al_sort.bam > out.out
получив результат, записанный в файл out.out.
Таким образом, на геном откартировалось 10867 ридов из 10868, т.е. не откартировался всего
один. Что значат остальные цифры непонятно.
(*) Определение среднего покрытия для экзона
С помощью команды:
samtools depth al_sort.bam > cover.out
был получен файл cover.out с вычисленными покрытиями для нуклеотидов. Я выбрала
нуклеотид в позиции 44717428 (покрытие 105). Экзон, содержащий этот нуклеотид (5), был найден
в GenomeBrowser. Он имеет границы 44,717,420-44,717,527 и принадлежит гену NSF.
Далее с помощью команды:
samtools depth -r chr17:44717420-44717527 al_sort.bam > cover.exon
был получен файл cover.exon, содержащий покрытия нуклеотидов нашего экзона. В
таблице есть полученные данные, а также вычисленное среднее
покрытие, которое составило около 95.5 (много).
3. Поиск SNP и инделей
Сначала был создан файл с полиморфизмами в формате .bcf с помощью команды:
samtools mpileup -uf chr17.fasta al_sort.bam -o snp.bcf
Полученный файл snp.bcf подан на вход другой команде:
bcftools call -cv snp.bcf -o snp.vcf
в результате чего был получен файл snp.vcf, содержащий список отличий
между референсом и ридами в формате .vcf. Всего было найдено 52 SNP
и 4 делеции. Качество SNP варьирует ~ в пределах от 3 до 225 (очень хорошее), среднее
значение — 67. При этом для 56 находок качество >20 (хорошее). Максимальное покрытие
составляет 63, а для целых 24 SNP покрытие составляет 1 рид [не совсем понимаю, какое
покрытие мы условно считаем хорошим]. В табл. 1 приведено описание для трех полиморфизмов из
этого файла.
Таблица 1. Описание трех полиморфизмов
| Координата |
Тип полиморфизма |
В референсе |
В ридах |
Глубина покрытия |
Качество ридов |
| 44788310 |
Замена |
G |
A |
74 |
221.999 |
| 79539193 |
Индель (делеция) |
Ttttattta |
Tttta |
10 |
122.467 |
| 44833088 |
Индель (делеция) |
ATTTTTTTTTTTTTTTTTTT |
ATTTTTTTTTTTTTTTT,
ATTTTTTTTTTTTTTTTT |
101 |
5.78792 |
4. Аннотация SNP
В этой части задания необходимо было с помощью программы
annovar
проаннотировать только полученные SNP (индели были вручную удалены в файле
snp_only.vcf). Использовались базы данных: refgene, dbsnp, 1000 genomes, GWAS,
Clinvar.
Сначала необходимо было из нашего файла snp_only.vcf получить файл, с которым может
работать программа annovar. Для этого использовалась команда:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput
Далее полученный файл использовался для аннотации SNP с помощью скрипта
annotate_variation.pl.
1) Аннотация по базе refgene:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr17.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
В результате было получено три файла: chr17.refgene.variant_function (содержит описание
всех SNP), chr17.refgene.exonic_variant_function (содержит описание SNP, попавших в
экзоны) и chr17.refgene.log (содержит информацию о процессе работы). В таблице 2
приведена общая информация о найденных SNP из файла chr17.refgene.variant_function. В
нем SNP разделены на несколько групп в зависимости от положения в геноме (интроны, экзоны,
UTR и т.д.). Также в этом файле указано, какой является замена: гетерозиготной или
гомозиготной. Таким образом, в область экзонов попало только три SNP, краткая информация о
которых приведена в таблице 3.
Таблица 2. Общая информация о найденных SNP
| intronic |
exonic |
UTR3 |
hom |
het |
| 44 |
3 |
5 |
34 |
18 |
Таблица 3. Описание трех полиморфизмов
| Координата |
Тип замены |
Ген |
В референсе -> в ридах |
Замена аминокислоты |
Глубина покрытия |
Качество ридов |
Изображение в IGV |
| 62007498 |
синонимичная |
CD79B |
A -> G |
C122C, C123C |
63 |
221.999 |
тык |
| 79589242 |
синонимичная |
NPLOC4 |
G -> A |
T53T |
104 |
221.999 |
тык |
| 79596811 |
синонимичная |
NPLOC4 |
C -> T |
P12P |
48 |
221.999 |
тык |
Как видно из таблицы, больше всего замен было найдено в интронах, а меньше всего — в
экзонах, что, в принципе, понятно: замены в кодирующих последовательностях могут привести к
замене аминокислоты в соответствующей белковой последовательности и, таким образом, функция
того или иного белка может быть нарушена. Замены же в некодирующих последовательностях не
подвергаются отбору, т.к. не кодируют белки. При этом все три замены нуклеотидов в экзонах
синонимичны, т.е. они не привели к замене аминокислоты в белке на другую.
Немного информации об упомянутых генах и белках, которые они кодируют:
- CD79B (Cluster of Differentiation 79B) — ген, кодирующий белок Ig-β,
входящий в состав белкового комплекса, образующего рецептор В-клетки, узнающий
антиген1;
- NPLOC4 (Nuclear protein localization protein 4 homolog) — ген, кодирующий
белок NPL4 homolog), который вместе с двумя другими белками образует комплекс, связывающийся
с убиквитинированными белками и обеспечивающий экспорт неправильно свернутых белков из ЭПС в
цитоплазму, где они затем подвергаются деградации протеасомой2. На NCBI также имеет второе название: ubiquitin recognition
factor3.
(*) Визуализация полиморфизмов
С помощью программы IGV
описанные полиморфизмы необходимо было визуализировать. Для этого в программе был открыт файл
al_sort.bam. Затем в строке поиска были указаны позиции моих полиморфизмов. Так как
изображения получились слишком большими, здесь я их выкладывать не буду; их можно открыть,
нажав на соответствующую ссылку в таблице выше (откроются в новой вкладке).
2) Аннотация по базе dbsnp:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr17.dbsnp -build hg19 -dbtype snp138 snp.avinput
/nfs/srv/databases/annovar/humandb/
В результате было получено 3 файла: chr17.dbsnp.hg19_snp138_dropped (SNP, имеющие
идентификатор rs, т.е. аннотированные в базе данных dbSNP), chr17.dbsnp.hg19_snp138_filtered
(SNP, не имеющие rs) и такой же файл chr17.dbsnp.log, как в предыдущем случае.
Таким образом, 46 SNP имеют аннотацию в упомянутой базе данных, а 6 — не имеют. При
этом все SNP без аннотации имеют низкое качество ридов, в то время как для SNP с rs качество
ридов сильно варьирует.
3) Аннотация по базе 1000 genomes:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr17.1000 -build hg19 -dbtype snp138 snp.avinput
/nfs/srv/databases/annovar/humandb/
Тут все аналогично: опять три файла с таким же содержимым, но только для базы 1000
genomes. В итоге в этой базе аннотировано 46 SNP, а не аннотировано — 6, как и в
предыдущем пункте. Частота варьирует от 0.000199681 (т.е. в крайней степени редко) до 0.979034
(очень часто). Примечательно, что SNP, входящие в экзоны (упоминались в аннотации по базе
refgene) встречаются часто (частота > 0.5).
4) Аннотация по базе GWAS:
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr17.gwas -build hg19 -dbtype gwasCatalog snp.avinput
/nfs/srv/databases/annovar/humandb/
Было получено 2 файла: chr17.gwas.log и chr17.gwas.hg19_gwasCatalog (содержит
SNP, для которых известно какое-то клиническое значение). В нашем случае было найдено 3
полиморфизма с аннотацией в GWAS: первый вызывает предрасположенность к раку
яичников и болезни Паркинсона, второй связан с ростом, а третий — с цветом глаз.
5) Аннотация по базе Clinvar:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr17.clincar -buildver hg19 -dbtype clinvar_20150629 snp.avinput
/nfs/srv/databases/annovar/humandb/
В итоге было получено 3 файлa. Не считая *.log, это файлы:
chr17.clincar.hg19_clinvar_20150629_dropped (содержит найденные аннотированные SNP) и
chr17.clincar.hg19_clinvar_20150629_filtered (содержит все остальные). В нашем случае
первый файл оказался пустым, т.е. в данном БД ни один из изучаемых SNP не аннотирован.
Все найденные SNP (учитывались только файлы *dropped) можно найти в общей таблице Excel:
pr13.xlsx.
Таблица 4. Сводная таблица со всеми командами
| Команда |
Что делает |
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr17.fastq chr17_out.fastq TRAILING:20 MINLEN:50 |
Принимает на вход файл с чтениями, в выходной файл записывает риды, длиной не менее
50 нуклеотидов (MINLEN:50), при этом с концов ридов отрезаны нуклеотиды с качеством ниже
20 (TRAILING:20) |
bwa index chr17.fasta |
Индексирует файл с референсной последовательностью |
bwa mem chr17.fasta chr17_out.fastq > align.sam |
Строит выравнивание ридов с референсной последовательностью и записывает их в файл
.sam |
samtools view align.sam -b -o align.bam |
Переводит файл с выравниванием в формат .bam (-b) и записывает все в файл,
указанный после -o |
samtools sort align.bam -T wtf.txt -o al_sort.bam |
Принимает на вход файл с выравниванием ридов с референсом и сортирует по координате
в референсе начала рида. Выходной файл после -o, в файл после -T записывается все, что
выдается в stdout |
samtools index al_sort.bam |
Индексирует файл с выравниванием |
samtools idxstats al_sort.bam > out.out |
Определяет, сколько ридов откартировалось на геном, записывая результаты в файл
(out.out) |
samtools depth al_sort.bam > cover.out |
Вычисляет покрытия для нуклеотидов и записывает результат в файл (cover.out) |
samtools depth -r chr17:44717420-44717527 al_sort.bam > cover.exon |
Вычисляет покрытия для нуклеотидов из указанного региона (-r) и записывает в файл
(cover.exon) |
samtools mpileup -uf chr17.fasta al_sort.bam -o snp.bcf |
Находит полиморфизмы и записывает их в файл (snp.bcf) |
bcftools call -cv snp.bcf -o snp.vcf |
Создает файл (после -o) с отличиями между референсом и рилами в формате .vsf
|
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_only.vcf -outfile snp.avinput |
Создает из входного файла файл, ск оторым может работать программа annovar |
perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr17.* -build hg19 -dbtype *DATABASE* snp.avinput
/nfs/srv/databases/annovar/humandb/ |
Создает файл с аннотацией SNP из входного файла (snp.avinout) по указанной базе
данных (*DATABASE*). |
© Mishchenko Polina 2016