Для выполнения задания я выбрала домен Globin (PF00042). Этот домен найден у 1761 видов в составе 4699 последовательностей белков, и всего для него известно 66 архитектур. Глобины представляют собой надсемейство гемосодержащих глобулярных белков, участвующих в связывании и/или транспортировке кислорода. В том числе в эту группу входят крайне распостраненные белки миоглобин и гемоглобин.
Ссылка на Globin.Для данного домена я скачала выравнивание (Jalview: File - Fetch Sequences - Pfam(Full) - PF00042), которое затем было покрашено (ClustalX, порог консервативности=30). Jalview отказался сохранять изображение выравнивания, поэтому привожу выравнивание в формате fasta или в качестве проекта.
Топ-5 архитектур ( по количеству последовательностей) представлены на рис.1

Я выбрала две архитектуры из списка. Архитектура 3 : число последовательностей - 147; домены - Globin, NAD_binding_1; Архитектура изображена на рисунке 2. Архитектура 4: число последовательностей - 55; домены - Globin x 2; Архитектура изображена на рисунке 3.


Сначала с помощью скрипта swisspfam_to_xls.py я отобрала последовательности с моим доменом из файла /srv/databases/pfam/swisspfam.gz, содержащего информацию об архитектуре всех последовательностей:
python swisspfam-to-xls.py -z -i swisspfam.gz -p PF00042 -o archi.xls
Для данной таблицы я создала сводную таблицу в которой столбцами являются разные домены, а строками - идентификаторы белков. Исследуемый домен я выделила зеленым, а второй домен из первой архитектуры - голубым. Затем для всех идентифекаторов белков была получена таксономия:
python uniprot-to-taxonomy.py -i pr11.txt -o tax.xls
Всю полученную информацию вы можете найти в .xlsфайле. На оновании анализа полученой сводки мною в качестве таксона был выбран тип Metazoa, а в качестве двух его подтаксонов - классы Ecdysozoa (E) и Chordata (C)
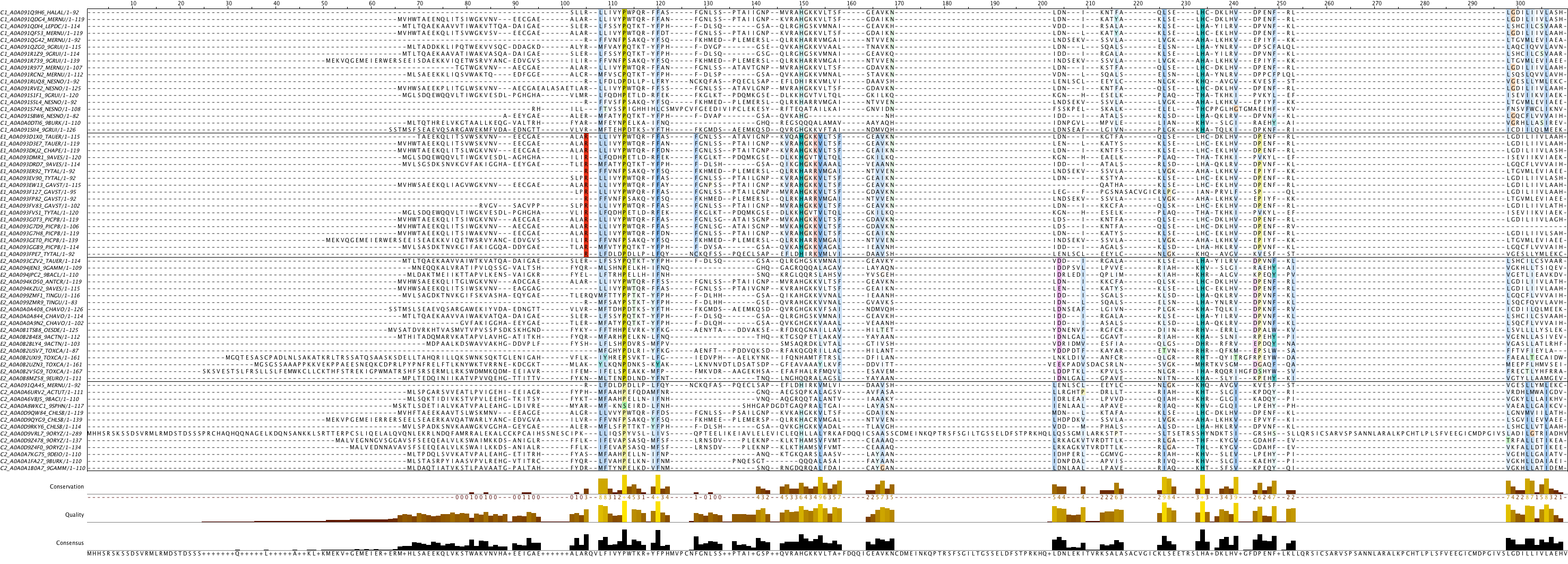
Затем я получила файл с идентификаторами выбранных мной последовательностей из каждого таксона(E1-Ecdysozoa c Globin, E2-Ecdysozoa c NAD_binding_1 и Globin, С1-Chordata c Globin, С2-Cordata c NAD_binding_1 и Globin).Затем с помощью скрипта filter-alignment.py я о ставила в выравнивании только выбранные мною последовательности:
python filter-alignment.py -i uniprot.fasta -m EC.txt -o my.fa -a "_"
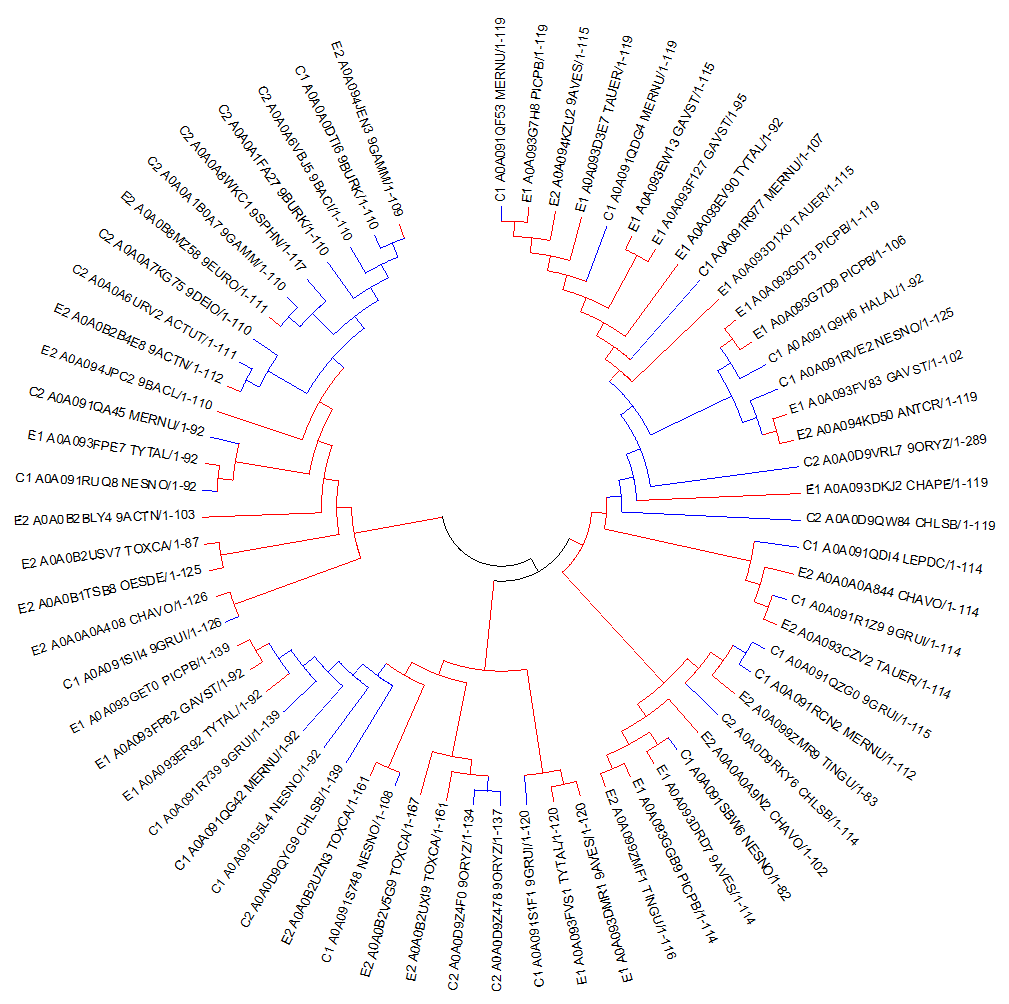
Далее к идентификаторам в полученном выравнивании я приписала выбранные обозначения для таксонов и архитектур. Также я подчистила выравнивание, выделила 4 группы по архитектуре и таксонам и раскрасила выравнивание Clustalx (30%). Полученное выравнивание представлено в .mfa формате, как проект, а так же изображено на рисунке(4) внизу