Сборка генома de novo
Получение чтений и общего файла с адаптерами
Для получения файла с ридами была использована следующая команда:
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/000/SRR4240360/SRR4240360.fastq.gz
Затем последовательности адаптеров были объединены в общий файл adapters.fa следующей командой:
cat /mnt/scratch/NGS/adapters/* >> adapters.fa
Очистка ридов с помощью trimmomatic
Затем была проведена очистка ридов с помощью программы trimmomatic. Сперва файл был разорхивирован и были удалены адаптерные последовательности при помощи следующей команды:
java -jar /usr/share/java/trimmomatic.jar SE SRR4240360.fastq trimmed.fastq -trimlog trim.log ILLUMINACLIP:adapters.fa:2:7:7
Файл с логом программы: здесь. Было удалено 41858 ридов из 8354632, что составляет 0,51% (осталось соответсвенно 99,49% ридов). Следующим шагом было проведено удаление с правого конца чтений нуклеотидов с качеством меньше 20, и последующее удаление чтений с длиной меньше 32 нуклеотидов. Команда представлена ниже:
java -jar /usr/share/java/trimmomatic.jar SE trimmed.fastq trimmed2.fastq -trimlog trim2.log TRAILING:20 MINLEN:32
Файл с логом программы: здесь. В результате работы программы осталось 7915474 (96,38%) ридов, было удалено 3,62%. В итоге по сравнению с изначальным файлом, вес которого составлял 872,8 МВ, вес файла после выполнения первой команды практически не изменился (868,1 МВ), вес файла после выполнения второй команды также слегка уменьшился (834,8 МВ). Исходя из этого можно сказать, что изначальные файлы были достаточно качественными и уже очищенными от адаптеров.
Запуск velveth
Для создания k-меров на основе ридов длиной 31 нуклеотид была введена следующая команда:
velveth velveth 31 -fastq -short trimmed2.fastq &> velvet.lo
Опция-fastq задаёт формат входного файла, -short говорит о коротких чтениях. На выходе получилась директория velveth с несколькими файлами в ней.
Запуск velvetg
Далее была запущена программа velvetg. Команда запуска представлена ниже:
velvetg velveth &> velvet.log
Файл с логом программы: здесь. Из этого файла узнаем значение параметра N50 - 43070. Также в результате работы программы были созданы файлы contigs.fa (в котором представлены контиги) и stats.txt (в котором представлена статистика по этим контигам).
Информация о самых больших контигах была получена с помощью команды:
sort -n -r -k 2 stats.txt | head
Ниже представлена информация о трёх самых длинных контигах.
| ID контига | Длина | Покрытие |
| 1 | 113474 | 33,53 |
| 5 | 83603 | 33,65 |
| 4 | 64155 | 35,85 |
Из аномально больших покрытий встречается покрытие в 134953 рида у контига 173, а из аномально маленьких покрытий встречается несколько покрытий равных 1.
Megablast для трёх самых длинных контигов
C помощью seqretsplit из файла contigs.fa были получены последовательности контигов с ID 1,4 и 5. После этого было произведено сравнение программой megablast (так как последовательности очень похожи) с референсным геномом. В Таблице 2 можно найти информацию о трёх произведённых выравниваниях.
| ID контига | E-value | Query Cover | Identity | Total Score | Ссылка на отчёт |
| 1 | 0.0 | 76% | 81% | 51702 | файл |
| 4 | 0.0 | 70% | 78% | 28200 | файл |
| 5 | 0.0 | 58% | 75% | 26995 | файл |
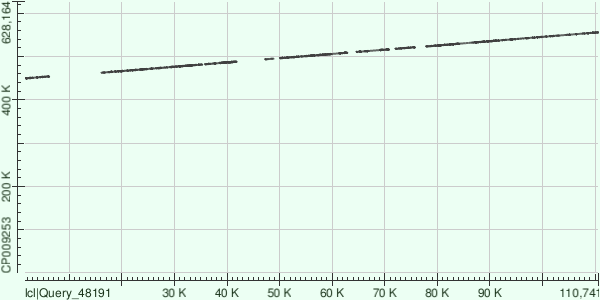
Контиг 1 соответствует референсному геному на участке с 528794 по 550219 нуклеотид. В лучшем выравнивании - 545 гэпов. (2%)
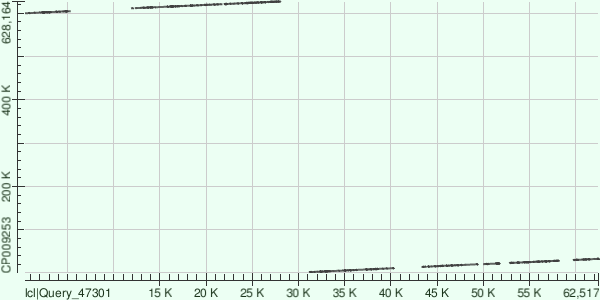
Контиг 4 соответствует референсному геному на участке с 2004 по 11103 нуклеотид.
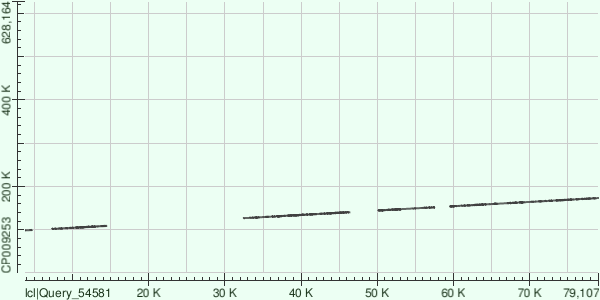
Контиг 5 соответствует референсному геному на участке с 127825 по 140555 нуклеотид. В лучшем выравнивании 548 гэп (4%).