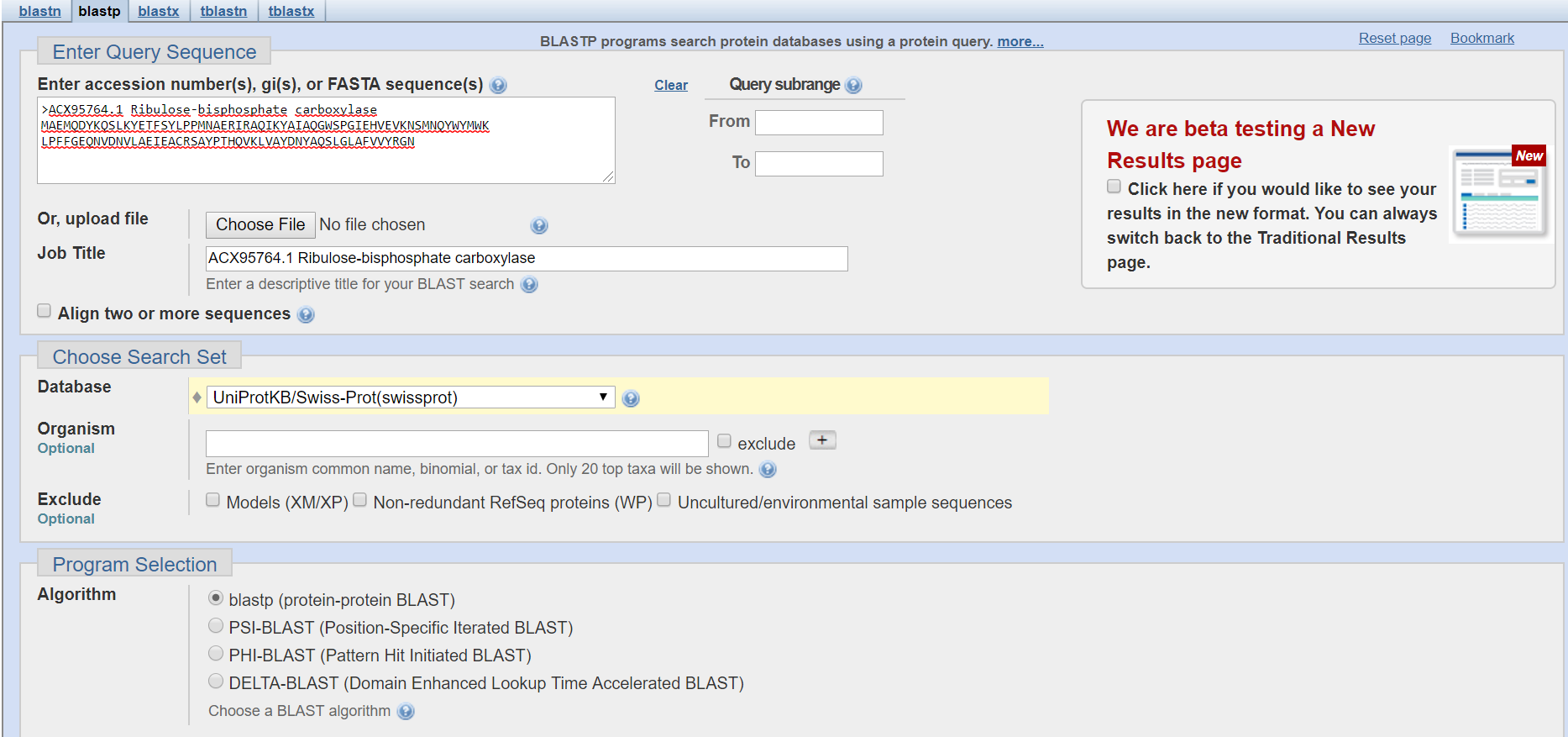
Параметры поиска в BLAST(1)
В поле вводится или загружается(кнопкой ниже) файл с последовательностью в формате FASTA или ID|AC белка.
С какой аминокислоты по какую учитывать при поиске.
Рабочее название последовательности, которое будет отображено в результатах поиска(ни на что не влияет).
Позволяет выполнять поиск по нескольким последовательностям.
Выбор базы данных.
Ограничивает поиск по указаному организму(ам), если поставить галочку в "exclude", то поиск будет осуществлятся по всей базе данных исключая указанные организмы.
Позволяет исключать из поиска некоторые последовательности:
- Models (XM/XP): последовательности с аннотациями, сделанными автоматически(конвеер).
- Non-redundant RefSeq proteins (WP): референсные геномы(исходные/эталонные).
- Uncultured/environmental sample sequences : последовательности полученные не из культур, прямиком из среды, в которой обитает организм.
Выбор алгоритма поиска.
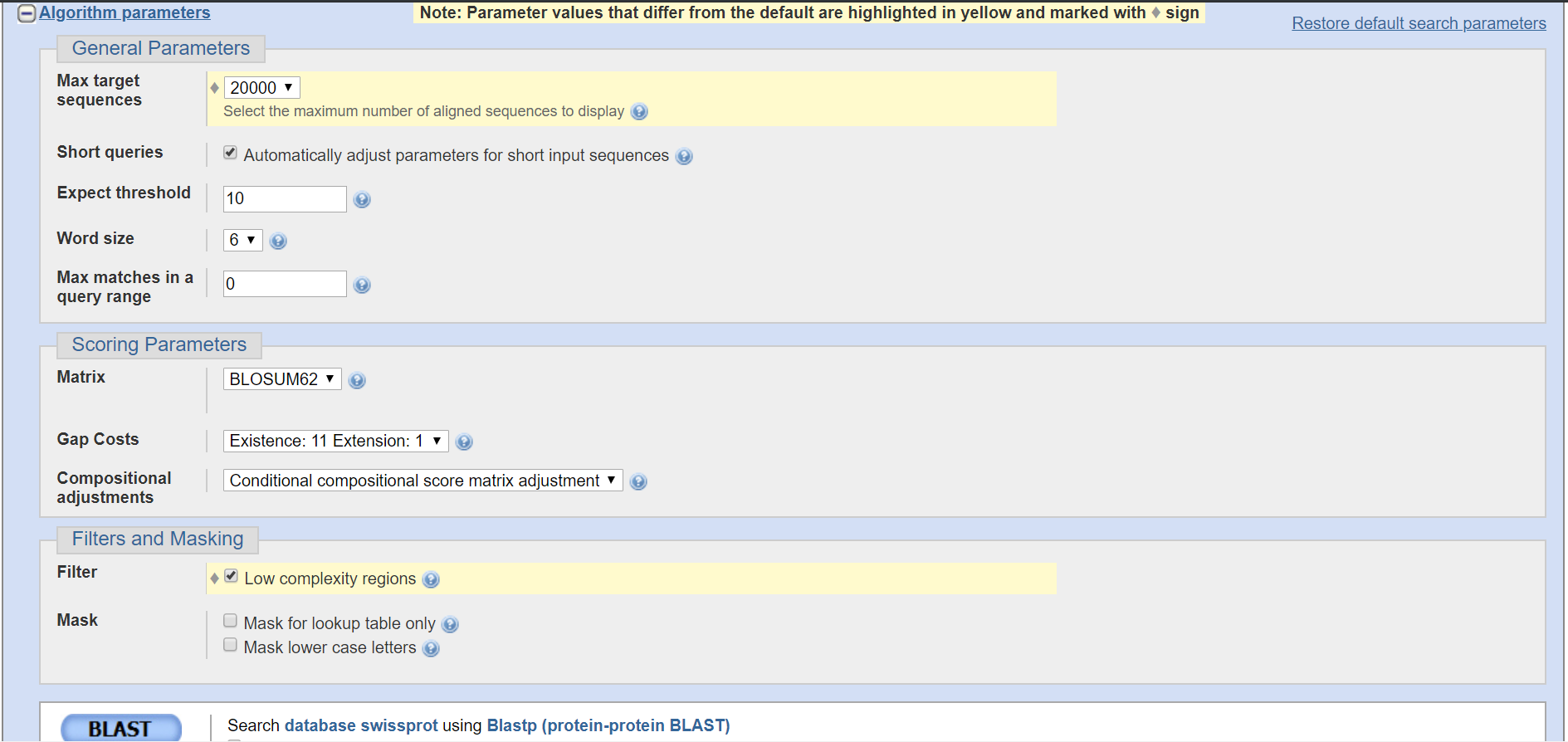
Параметры поиска в BLAST(1)
Количество последлвательностей, отображаемое по заданному запросу, я задал 20000, чтобы не упустить ни одной, так как число реально найденных последовательностей может превышать заданное ограничение.
Автоматически подбирает параметры для коротких последовательностей, если таковые находятся, я отметил на всякий случай для улучшения поиска и среди коротких.
Ожидаемое значение E-value, то есть его допустимый максимум: чем он меньше, тем более искомая последовательность должна быть увесистие в битах, поэтому я указал 10, чтобы оставить несколько низкопроцентных, может быть они будут интересны с точки зрения других своих значений.
Длина куска последовательности по которой проводиться первичный поиск, то есть сначала выравниваются куски по 6 аминокислот(в моем случае), своего рода локальные выравнивания. Я указал 6, поскольку считаю, что данная размерность дает наиболее точные результаты, и отметает случайные совпадения, поскольку она максимальна.
Ограничивает количество совпадений между искомыми и исходной последовательностью(integer). Если указан "0", то нет ограничений: я рассматриваю последовательности с любым количеством совпадений.
Матрица по которой определяется вес последовательности. Я выбрал "BLOSUM62", поскольку она была составлена на основе банка данных(размер и банк не помню, но верить этой статистике можно) и при составлении уситывались последовательности со схожестью в 62% и выше.
Штраф за GAP в матрице. Поскольку GAP серьезно меняет последовательность и функции белка, то стоит серьезно за него наказывать, но неслишком, чтобы не упустить возможных интересных исключений.
Позволяет масштабировать E-value относительно найденной выборки из последовательностей, то есть считает более относительные "Е", вместо глобальных, чисто по матрице.
Позволяет не выравнивать куски последовательностей с низкой композиционной ценностью, что означает, что будут найдены наиболее интересные с исследовательской точки зрения последовательности.
-Mask for lookup table only
Поиск только по таблице поиска, без исключений: поиск по заранее составленной таблице с первичными выравниваниями (в моем случае по 6 аминокислот).
-Mask lower case letters
Поиск будет производиться по той части последовательности, которая записана маленькими(lower case) символами.
Параметры поиска в BLAST(2)
Вот таблица составленная автоматически по результатам поиска.
В этом выравнивании представлены некоторые потенциальные гомологи, с лучшим покрытием, разнообразными E-value, и еще 2 последовательности с конца списка выдачи blast для сранения.

Выравнивание предполагаемых гомологов
Как видно из картинки, наиболее ярко выраженными гомологами я считал те последовательности, у которые составляют выделенной мной на картинке большой блок, тем не менее у них и без того хватает консервативных колоннок.
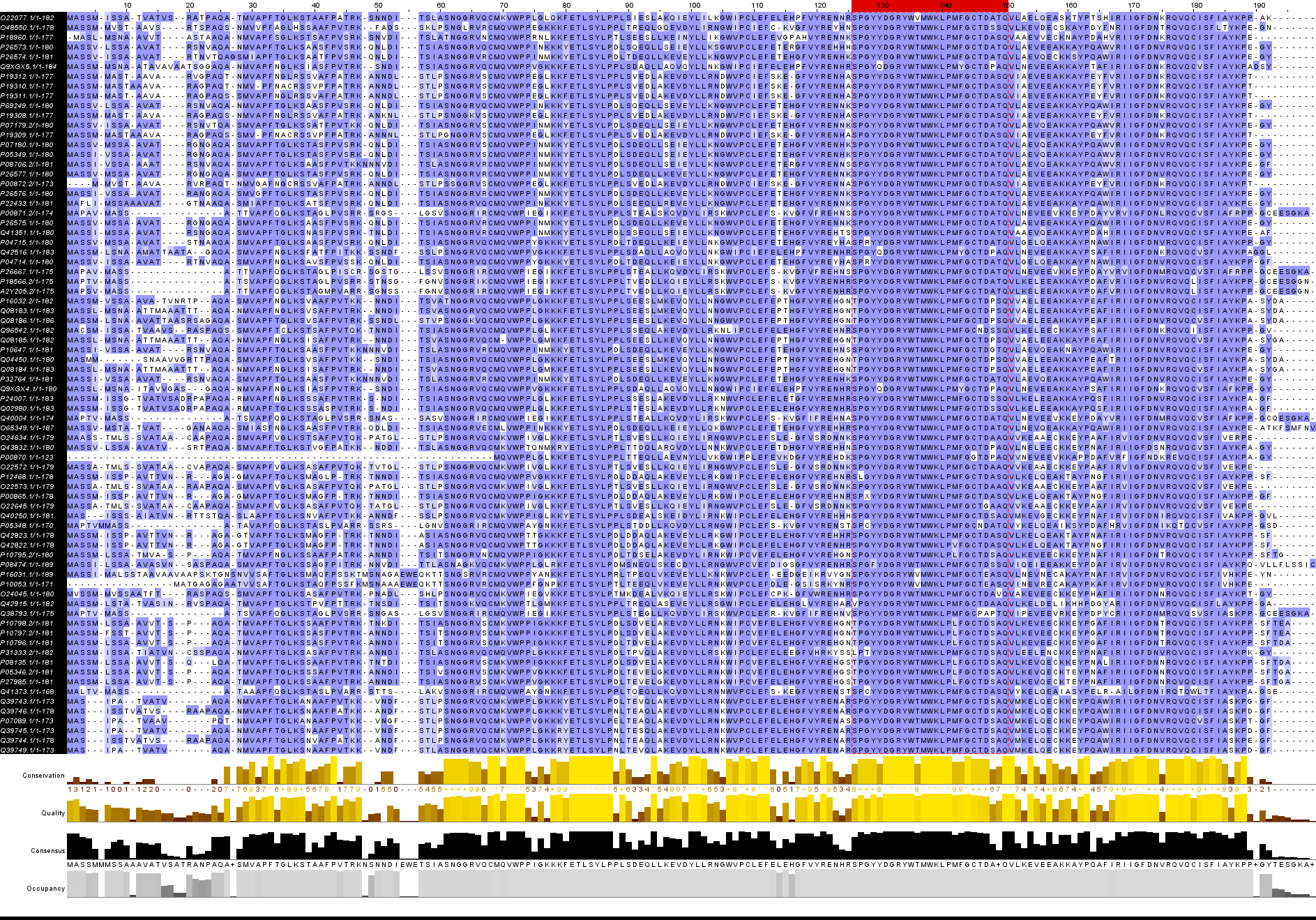
Выравнивание точных гомологов
1)Делеция в (1)
2)Делеция в (2)
3)Делеция в (2)
4)Делеция в (1)
5)Делеция в (1)
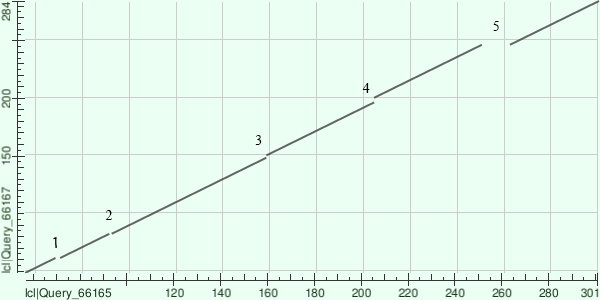
Карта локального сходства:
по оси абсцисс F4RBD6_MELLP (2),
по оси ординат A0A0D2WL23_CAPO3(1)
Фраза: "phantom thiefs wanna steal your heart"
1) При ожидаемом E-value = 100000 нашлась одна последовательность: ID: P65072.1(как бы высоко я его не поднимал, только одна).
2) При Word Size = 2 количество последовательностей достигло максимума(20000), но при этом есть последовательности с полным покрытием и не одна (например AC: Q9W0R6), а также минимальное E-value = 16.(при Word size = 3 мало что поменялось)
Выводы: Моя игра с параметрами еще раз подтвердила выбор мной параметров при первом поиске: если указать их такими, как это сделано в этом блоке выше, то в результате вылезет куча всякого мусора.