Задание 1
1. Несколько файлов в формате fasta собрать в один файл
Для этого список названий из файлов помещается в файл list, помещение файлов в один осуществляется с помощью команды seqret @list new.fasta
2. Один fasta-файл разделить на несколько
Возьмем файл new.fasta из предыдущего упражнения. Команда для его разделения:seqretsplit new.fasta. Данная команда выдаст новые файлы в формате название_последовательности.fasta.(1, 2, 3, 4
3. Из файла с хромосомой в формате .gbk вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле.
Возьмем геном герпеса (genbank:JN555585), создадим файл sequence, содержащий три CDS:
echo -e "genbank:JN555585[513:1259]\ngenbank:JN555585[9338:10012]\ngenbank:JN555585[10991..11665]">sequence
После чего получим последовательности:
seqret -sequence @sequence -outseq herpes.fasta
4. Транслировать несколько последовательностей в файле fasta в указанном генетическом коде.
Команда: transeq new.fasta -table 0 newp.fasta. Так как в задании не указан конкретный генетический код, то взят стандартный. Результат
5. Транслировать несколько последовательностей в шести рамках считывания:
transeq new.fasta -frame 6 newp6.fasta (newp6.fasta)
6. Перевести выравнивание из fasta-формата в msf-формат:
seqret -sequence align.fasta -outseq msf:align.msf (Исходник, Результат)
7. Вывести в stdout число совпадающих букв между второй последовательностью и всеми остальными (на выходе только ID и число)
infoalign -refseq 2 -only -name -idcount -sequence align.msf stdout
8. Перевести аннотацию особенностей из формата .gb в табличный формат .gff
featcopy -feature plasmid.gb -outfeat plasmid.gff (Исходник, Результат)
9.Из файла в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product).
extractfeat -type 'CDS' -describe 'product' -sequence plasmid.gb -outseq extracted.fasta (Исходник, Результат))
10. Перемешать буквы в данной нуклеотидной последовательности; (*) проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите с порогом E = 10 - по умолчанию).
shuffle extracted.fasta > random.fasta (Исходник, Результат)
Если взять первую последовательность из полученных случайных, то находится только 2 последовательности с e-value 4.9, т.е., находки однозначно недоставерны.
11. Найти частоты кодонов в данной кодирующей последовательности
cusp extracted.fasta extracted.cusp (Результат)
12. Найти частоты динуклеотидов с некоторой последовательности и сопоставить с ожидаемыми. Для этого взята первая последовательность из файла extracted.fasta
compseq -sequence ex1.fasta -calcfreq -word 2 -outfile ex1.compseq (Исходник, Результат)
Максимальные отклонения от ожидаемой частоты динуклеотидов наблюдаются у TA и TG, -35% и +40% отклонения от ожидаемой частоты соответственно.
13. Выровнять кодирующие последовательности соответственно выравниванию белков - их продуктов.
tranalign n.fasta p.fasta tranalign.fasta (Исходные последовательности нуклеотидов, Исходное выравнивание, Результат)
Задание 2
Для выполнения задания были взяты последовательности Escherichia fergusonii ATCC 35469 chromosome и Escherichia albertii DNA, complete genome, strain: EC06-170. Получившееся покрытие: 75%, сходство гомологичных участков: 94%.
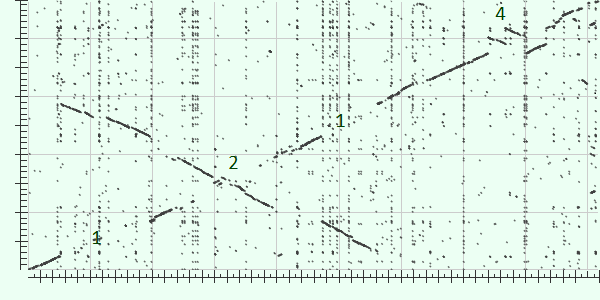
Рис. 1. Матрица сходства для взятых геномов
В местах, отмеченных как 1, видно появление инвертированных повторов, 2 - инверсия. Отметка 4 более интересна - видно транслокацию, которая претерпела инверсию и дупликацию в одном из геномов.
Для уменьшения шума на матрице E-value было ограничено значением 1e-09 и размер слова был увеличен до 15.