Подготовка чтений
Для сборки de novo генома бактерии Buchnera aphidicola str. Tuc7, код доступа SRR4240360, нужно сначала скачать файл с чтениями и подготовить файл с адаптерами, которые будут в дальнейшем вырезаться.
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/036/SRR4240360/SRR4240360.fastq.gz
cat ../../adapters/* > adapters.fa
Примечание: рабочая директория /mnt/scratch/NGS/kprokapovichd/pr14
Затем с помощью программы Trimmomatic триммируем чтения, удаляем нуклеотиды с конца, у которых качество ниже 20, и оставляем только те чтения, у которых длина 32 и более нуклеотида.
TrimmomaticSE -phred33 SRR4240360.fastq.gz SRR4240360_trim.fastq.gz ILLUMINACLIP:adapters.fa:2:7:7 TRAILING:20 MINLEN:32 2> logs.txt
В логах смотрим, что было удалено 339158 (4.11%) чтений, а размер файла изменился на 10М по сравнению с нетриммированным.
Собираем воедино
Для сборки генома нужно подготовить сначала хэш-таблицу с k-мерами (в нашем случае длиной 31):
velveth . 31 -short -fastq.gz SRR4240360_trim.fastq.gz
Получаем два файла: Roadmaps и Sequences.
Затем собираем контиги из этих k-меров:
velvetg .
Выдача такой программы дает аж 5 файлов, но нас интересует contigs.fa и Log. Из Log узнаем, что N50 = 43070, а из файла с контигами посмотрим самые длинные контиги:
grep -e "^>" contigs.fa | tr '_' '\t' | sort -k 4 -nr | head
Для более детального анализа вместо head я перенаправил выдачу в файл contigi.tsv, в excel легче всего найти медиану.
Кстати, из этой же таблицы можно увидеть, что L50 = 5. Там я нашел медиану (78), и 37 контигов оказлось больше медианы в 5 и более раз (максимум в ~1072 раза), а самый короткий контиг составляет 0,4 от медианной длины контига.
Самые длинные контиги NODE 1, 5 и 4 (в порядке убывания):
- NODE1: длина - 113 474, покрытие - 33.525459
- NODE5: длина - 83 603, покрытие - 33.646065
- NODE4: длина - 64 155, покрытие - 35.847324
Посмотрим, что это вообще за контиги повнимательнее...
Анализ
Для начала отберем нужные последовательности в отдельные файлы:
sed -n "/>NODE_1_/,/>NODE_2_/p; />NODE_2_/q" contigs.fa > node1.fa
sed -n "/>NODE_5_/,/>NODE_6_/p; />NODE_6_/q" contigs.fa > node5.fa
sed -n "/>NODE_4_/,/>NODE_5_/p; />NODE_5_/q" contigs.fa > node4.fa
А ещё нужно выбрать другой штамм нашей бактерии, на сайте NCBI найдем по видовому названию сборки, возьмем референсный Buchnera aphidicola str. Sg (Schizaphis graminum), берем RefSeq ID, ищем в RefSeq AC:
RefSeq AC: NC_004061
Приступаем к выравниванию с помощью megablst.

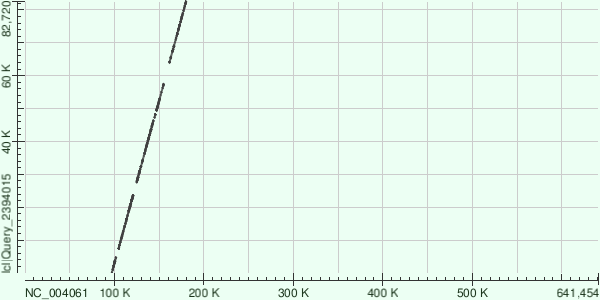
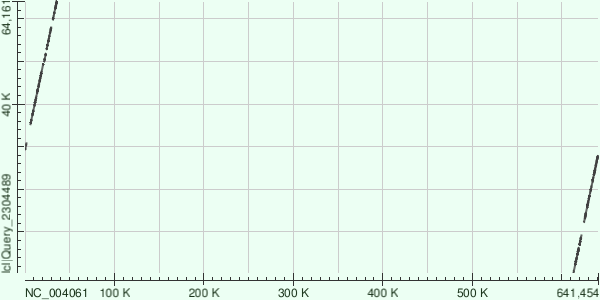
Как видно, контиги очень хорошо ложатся на хромосому. В некоторых местах есть делеции, NODE4 ложится на одно место, так как хромосома этой бактерии кольцевая.
NODE1: координаты хромосомы 458381-567722
NODE5: координаты хромосомы 96970-176511
NODE4: координаты хромосомы 14-35607 и 612905-640402 (т.е. по сути 612905-35607 с "перескоком", т.к. хромосома кольцевая)
К сожалению я не смог посчитать общее количество гэпов и snp, т.к. у меня контиг выравнивался по частям((( Но при быстром просмотре разных участков гэпов примерно 1-2% от общего количества позиций в выравнивании, а идентичность 70-80%.