В предыдущем задании были отобраны
некоторые бактерии, для которых было составлено соответствующее филогенетическое дерево:
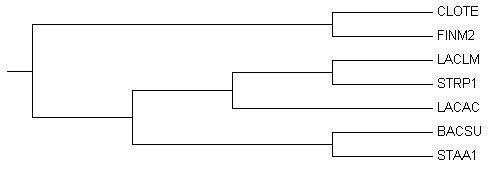
Далее с помощью таксономического сервиса NCBI
были определены таксоны, к которым эти бактерии относятся:
| Название |
Мнемоника |
Тип |
Класс |
Отряд |
Семейство |
| Bacillus subtilis |
BACSU |
Firmicutes |
Bacilli |
Bacillales |
Bacillaceae |
| Clostridium tetani |
CLOTE |
Firmicutes |
Clostridia |
Clostridiales |
Clostridiaceae |
| Finegoldia magna |
FINM2 |
Firmicutes |
Clostridia |
Clostridiales |
Clostridiales Family XI |
| Lactobacillus acidophilus |
LACAC |
Firmicutes |
Bacilli |
Lactobacillales |
Lactobacillaceae |
| Lactococcus lactis |
LACLM |
Firmicutes |
Bacilli |
Lactobacillales |
Streptococcaceae |
| Staphylococcus aureus |
STAA1 |
Firmicutes |
Bacilli |
Bacillales |
Staphylococcaceae |
| Streptococcus pyogenes |
STRP1 |
Firmicutes |
Bacilli |
Lactobacillales |
Streptococcaceae |
Из таблицы видно, что 5 бактерий относятся к классу Bacilli, а оставшиеся две к Clostridia. При этом можно
заметить, что каждая бактерия выделяет отдельный таксон, кроме Lactococcus lactis и Streptococcus pyogenes, которые
относятся к семейству Streptococcaceae.
Из предложенного в задании из
предложенного списка функций белков была выбрана функция «Фактор инициации трансляции 2», IF2.
Введя в Uniprot через точку с запятой идентификаторы вида IF2_*****, где ***** - мнемоника соответствующей бактерии,
получим из Swiss-Prot последовательности белков с нужной нам функцией из отобранных бактерий.
Далее полученные 7 последовательностей, объединим в один fasta-файл
(названия последовательностей для удобства совпадают с мнемоникой соответствующей бактерии).
С помощью алгоритма Muscle with Defaults в JalView получим нужное выравнивание и раскрасим по проценту Identity,
а также разделим на блоки по 100 с помощью Wrap:

Выравние в jar и fasta форматах.
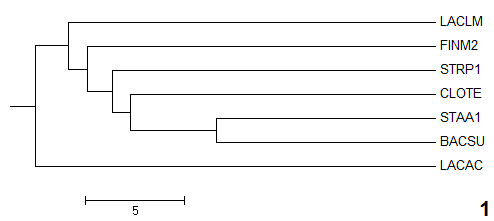
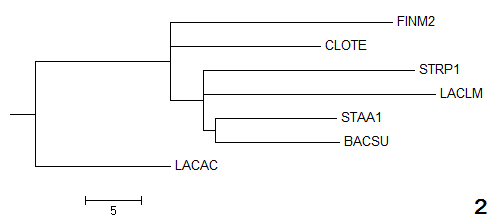
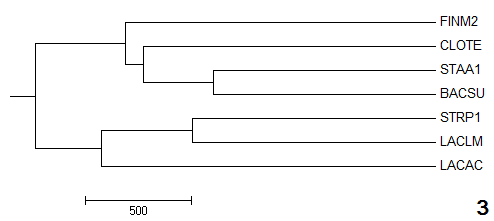
В меню Calculate -> Calculate Tree jalView доступны 4 метода реконструкции деревьев. Для каждого из
методов был создан соответствующий newick-файл. Визуализиуем данные деревья с помощью Mega:
|
Average Distance |
Neighbour Joining |
| % Identity |
 |
 |
| BLOSUM62 |
 |
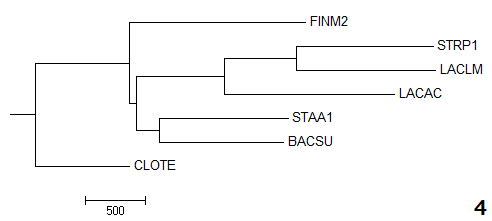 |
Сравним топологию полученных деревьев с исходным. Ветви, отсутствующие на правильном дереве,
но присутствующие на на реконструированных, и наоборот, отметим в таблице ниже:
|
|
Только на правильном
|
Только на реконструированном
|
| 1 |
{CLOTE,FINM2}vs{LACLM,STRP1,LACAC,BACSU,STAA1}
{LACLM,STRP1}vs{LACAC,BACSU,STAA1,CLOTE,FINM2}
{LACLM,STRP1,LACAC}vs{BACSU,STAA1,CLOTE,FINM2}
|
{BACSU,STAA1,CLOTE}vs{LACLM,STRP1,LACAC,FINM2}
{BACSU,STAA1,CLOTE,STRP1}vs{LACLM,LACAC,FINM2}
{BACSU,STAA1,CLOTE,STRP1,FINM2}vs{LACLM,LACA}
|
| 2 |
{LACLM,STRP1,LACAC}vs{CLOTE,FINM2,BACSU,STAA1} |
{BACSU,STAA1,LACLM,STRP1}vs{CLOTE,FINM2,LACAC} |
| 3 |
{CLOTE,FINM2}vs{LACLM,STRP1,LACAC,BACSU,STAA1} |
{BACSU,STAA1,CLOTE,FINM2}vs{LACAC,LACLM,STRP1} |
| 4 |
{LACLM,STRP1,LACAC,BACSU,STAA1}vs{CLOTE,FINM2} |
Соответствующая ветвь не была найдена |
Импортируйте fasta-файл с выравниванием в программу Mega: при импорте выберем "Analyze". Затем реконструируем
дерево методом Maximum Parsimony (в меню Phylogeny). Укореним полученное дерево:
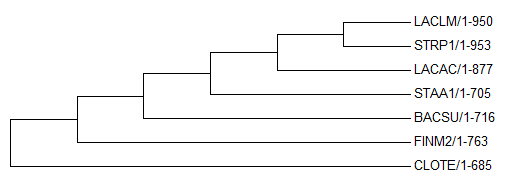
Полученное дерево также отличается от исходного.
Ни один из методов реконструкции не дал правильного дерева,
все полученные деревья воспроизводят правильное лишь до некоторой степени,
В любом cлучае, не стоило ожидать большего от реконструкции всего лишь по одному белку.