Выбор структуры белка
В качестве модели невозможно было выбрать структуру белка, изучаемого на первом курсе - UvrABC system protein B, так как она не удовлетворяла критериям.
В результате была использована последовательность пиродоксаминкиназы (PDXY_ECOLI), вернее, ее стркутура:
PDB ID: 1VI9 (Разрешение - 1.96Å)
Данный белок соответствует следующим условиям:
- Белок есть в базе данных EDS, то есть для него доступны экспериментальные данные. Файл, описывающий электронную плотность, был скачан с этого сайта.
- Для белка известно более 5 структурных гомологов с RMSD от 0.8 до 3.0 ангстрем и N_align от 50 до 90% числа аминокислотных его остатков.
(Проверка осуществлялась с помощью PDBeFold) RMSD (Root Mean Square Deviation).
Для визуализации структуры и электронной плотности была использована программа PyMol. Скачанные файлы были загружены в PyMol с помощью следующих команд:
load 1vi9.pdb, 1vi9
load 1vi9.omap, 1vi9_map
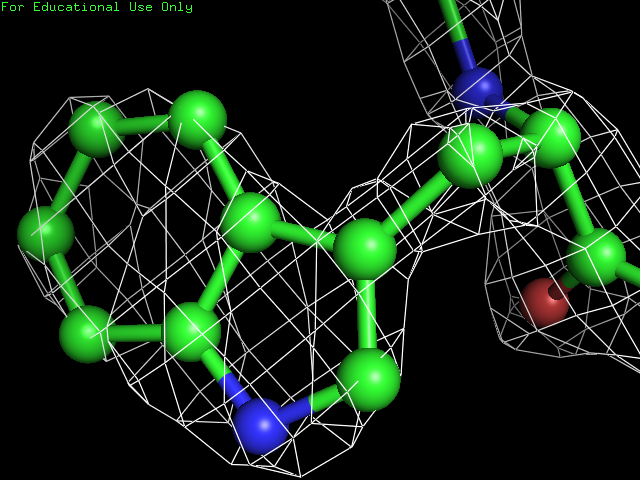
Построение изображения ЭП
Для получения изображений электронной плотности вокруг полипептидной цепи для уровней электронной плотности 1.0 σ, 1.5 σ и 2.0 σ на расстоянии 2 ангстрема использовались, соответственно, команды:
isomesh backbone_map, 1vi9_map, 1, backbone, carve=2
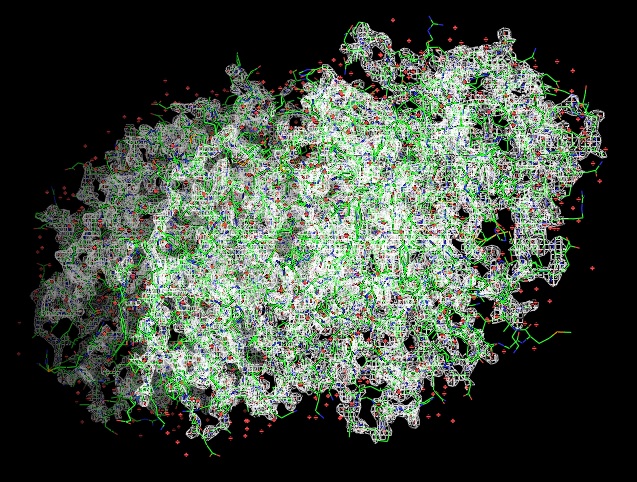
isomesh backbone_map, 1vi9_map, 1.5, backbone, carve=2
isomesh backbone_map, 1vi9_map, 2, backbone, carve=2
Полученные изображения приведены ниже (увеличиваются по щелчку):
Подрезка 1 σ Подрезка 1.5 σ Подрезка 2 σ 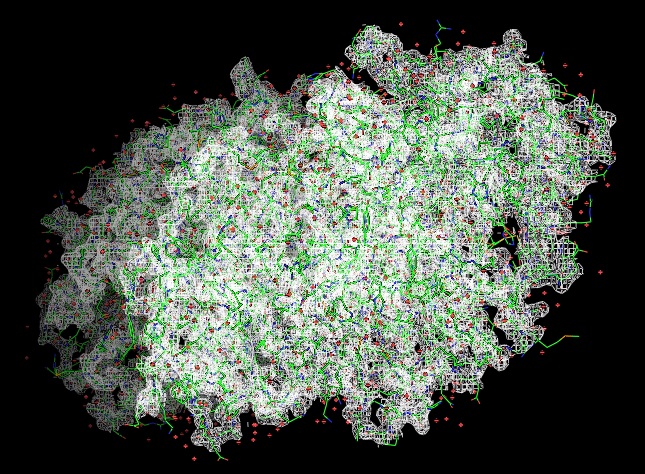

Построение изображения ЭП вокруг аминокислотных остатков
Для дальнейшего анализа были выбраны триптофан, лейцин и лизин.
С помощью команд (пример) в PyMol были получены следующие изображения (увеличиваются по щелчку):
Аминокислота\Уровень подрезки 1σ 1.5σ 2σ Тирозин 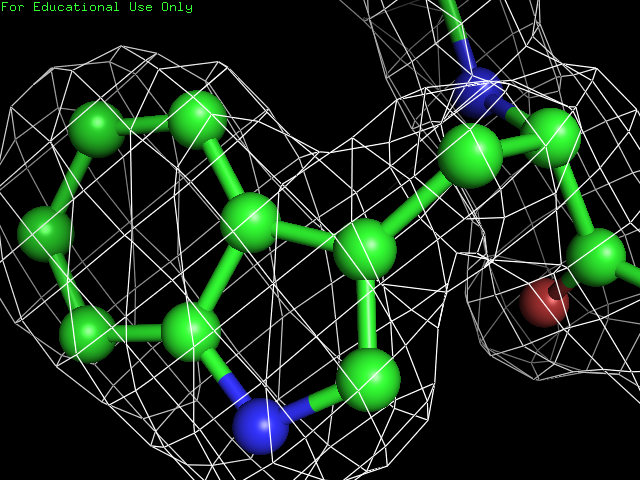
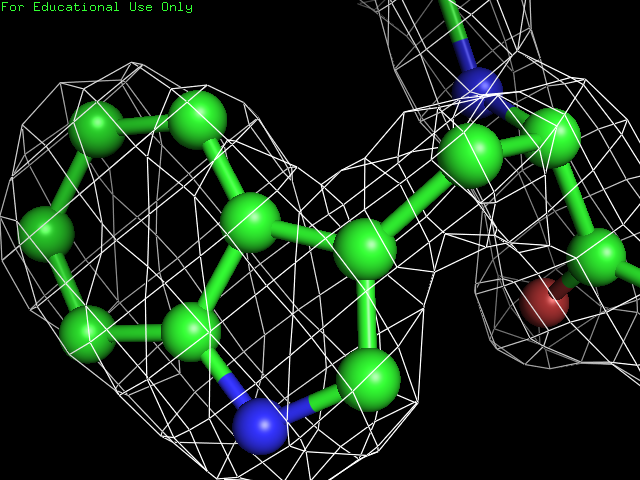
Лейцин 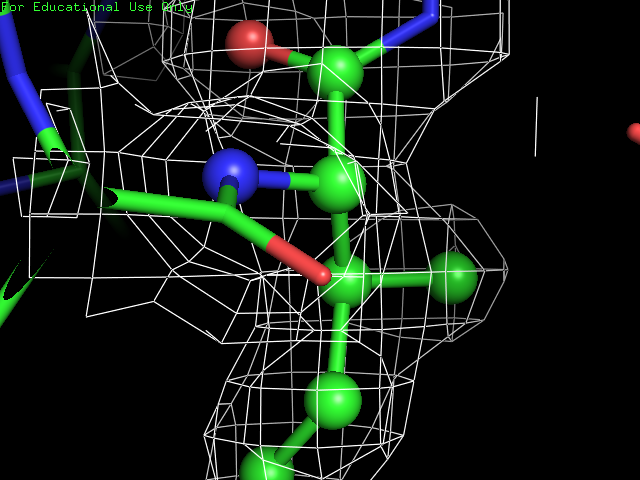
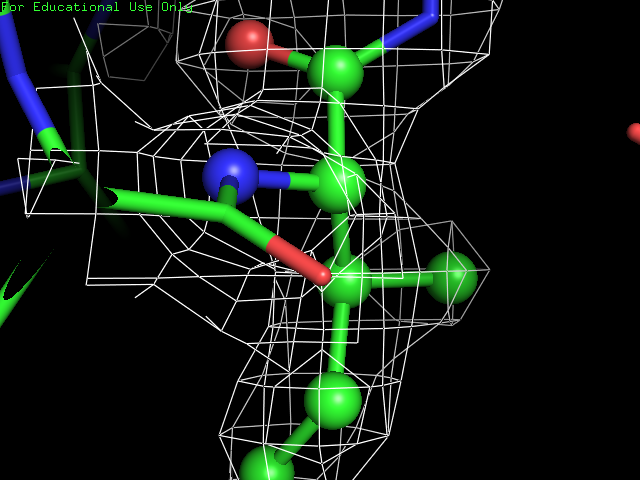
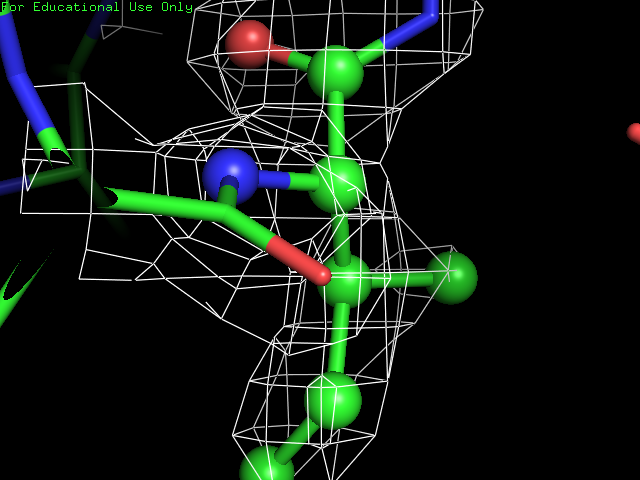
Лизин 
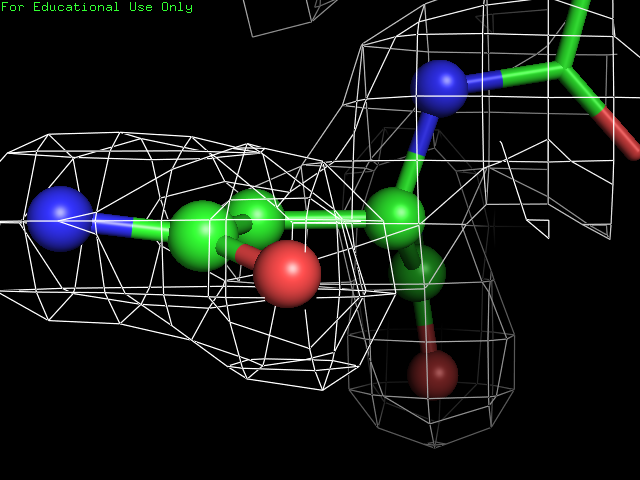
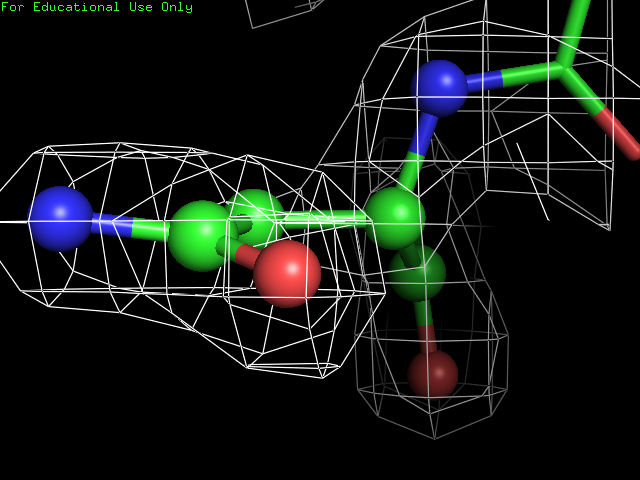
В целом, разрешения структуры достаточно для того, чтобы наблюдать большинство атомов боковых групп аминокислотных остатков.