Отчет по EMBOSS
Заданиe 1. Поиск организма по фрагменту нуклеотидной последовательности
Дан фрагмент нуклеотидной последовательноcти. C помощью megablast нужно найти полную последовательность генома бактерии или археи, откуда был взят этот фрагмент. Используем поиск в базе refseq_genomic. Программа выдала совпадение с геномом гипертермофильного симбионта Nanoarchaeum equitans. Исходный фрагмент распологается в геноме в координатах 1145..1444 (рис.1).
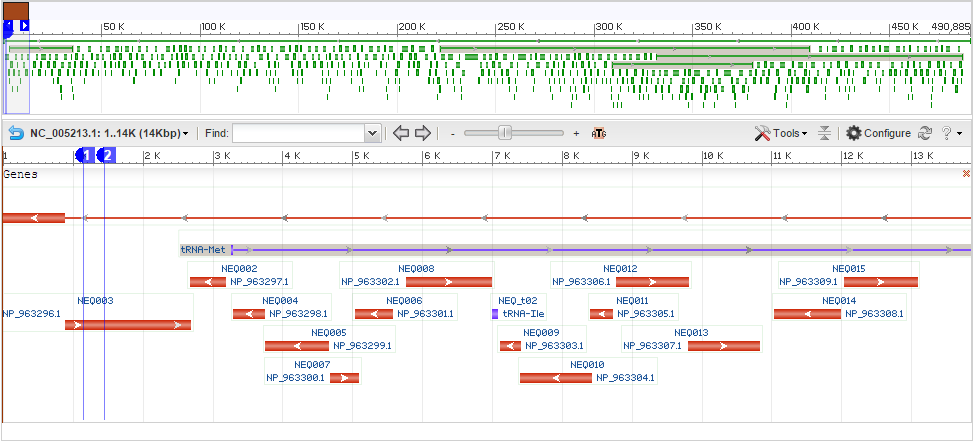
Как видно, фрагмент 1-2 находится внутри кодирующего участка. А именно, этот участок кодирует белок NP_963296.1 с неизвестной функцией:
CDS: NP_963296.1 Title: hypothetical protein Comment: COG1201: Lhr-like helicases; IPR001410: DEAD/DEAH box helicase; IPR001472: Bipartite nuclear localization signal; IPR001650: Helicase C-terminal domain; IPR003593: AAA ATPase superfamily Location: 883..2,691
Заданиe 2. Поиск гомолога белка человека в слоне
Будем рассматривать аннотированные (SwissProt) белки человека, AC которых начинается с букв "BE". Для того, чтобы найти их, использовалась такая команда: infoseq sw:be*_human -only -name -desc -out file_name.txt.
Возьмем белок BECN1_HUMAN. Beclin-1 играет важную роль в аутофагии, участвует в защите организма от вирусов. С помощью программы seqret получена последовательность белка.
Теперь нужно найти гомологов этого белка у довольно близкого в эволюции организма — африканского слона Loxodonta africana. С помощью сервиса сайта ENA spliced translated nucleotide search были получены следующие результаты:
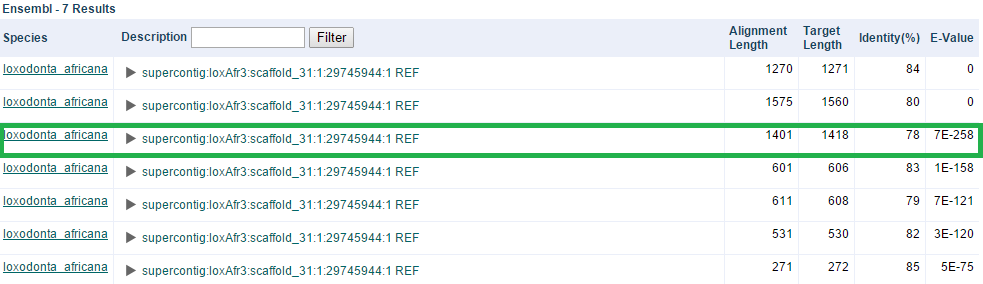
Как известно, у лучших находок значение E-value самое маленькое. Так что более достоверной находкой можно признать третью из представленных. Ее длина выравнивания 1401, identity 78%, e-value 7E-258, координаты в геноме слона 24402367-24403784, интронов нет.
Заданиe 3. Поиск некодирующих последовательностей программой BLAST
В этом задании будет рассматриваться нуклеотидная последовательность одной из молекул тРНК археи Aeropyrum pernix. Необходимо найти гомологов этой тРНК среди белков организмов того же порядка (Desulfurococcales). Это можно сделать тремя способами:
1. алгоритмом megablast;
2. алгоритмом blastn с параметрами по умолчанию;
3. алгоритмом blastn с длиной затравки 7, match/mismatch scores 1/-1(максимально чувствительные параметры, доступные на сайте)
Результаты отражены в таблице:
| Алгоритм | Число находок с E-value <0.001 |
| megablast | 3 |
| blastn по умолчанию | 13 |
| blastn с длиной слова = 7, match/mismatch = 1/-1 | 15 |
"Затравка" — длина участка, который инициирует выравнивание с какой-то последовательностью. В общем, результаты предсказуемы, ведь чем короче длина инциирующего участка и меньше штраф за совпадение/несовпадение аминокислот, тем больше найдется подходящих последовательностей.