Алгоритмы и программы множественного выравнивания. Базы гомологичных доменов.
Алгоритм сравнения разных выравниваний одних и тех же последовательностей
Программа принимает на вход 2 файла с выравниваниями в fasta-формате, а также название файла, в который будет записан список координат одинаково выровненных колонок в обоих выравниваниях (i,j). Программа выдает в stdout длину обоих выравниваний, процент выровненных колонок от длины каждого из выравниваний, координаты блоков идущих подряд одинаково выровненных колонок, а также создает файл со списком координат (i,j), упомянутый выше. Информацию о программе можно получить командой: python alis.py -h
Сравнение выравниваний одних и тех же последовательностей тремя разными программами
Выбранные программы: MAFFT, MUSCLE, T-Coffee: сравнение MAFFT с MUSCLE и MAFFT с T-Coffee. Я решила сравнить выравнивания белков – гомологов кинурениназы из практикума 10. Обе пары выравниваний не отличались по длине, доле одинаково выровненных колонок и координатам выровненных блоков. Различия есть только в невыровненных колонках, если искать визуально. Получается, если последовательностей мало, и они очень похожи, выравнивания, полученные разными алгоритмами, практически не отличаются. В качестве набора сильнее отличающихся последовательностей я выбрала белки (Methyl-coenzyme M reductase beta subunit) с разными доменными архитектурами, которые рассматривались в практикуме 11.
Ссылки на проекты Jalview: MAFFT MUSCLE T-Coffee
Результаты сравнения: MAFFT с MUSCLE: ali_1.len: 487 ali_2.len: 483 %ali.col1: 16.43% %ali.col2: 16.56% Список блоков одинаково выровненных колонок: (11-67) = (10-66) – length: 57 (107-129) = (107-129) – length: 23 В эти блоки входят все одинаково выровненные колонки. MAFFT с T-Coffee: ali_1.len: 487 ali_2.len: 486 %ali.col1: 16.63% %ali.col2: 16.67% Список блоков одинаково выровненных колонок: (11-68) = (10-67) – length: 58 (107-129) = (110-132) – length: 23 В эти блоки входят все одинаково выровненные колонки.
Можно заметить, что результаты выравнивания программой MAFFT сильнее похожи на результаты выравнивания программой T-Coffee, чем программой MUSCLE. MAFFT и T-Coffee: выравнивания почти одинаковы по длине (487 и 486), в начале есть очень длинный блок одинаково выровненных колонок, который отличается по координатам в каждом выравнивании только на 1 колонку. MAFFT и MUSCLE: выравнивания отличаются по длине на 4 колонки. В начале также есть длинный блок одинаково выровненных колонок. Второй такой блок одинаковой длины есть в обоих сравнениях выравниваний, однако при сравнении выравниваний, полученных MAFFT и T-Coffee, он отличается по координатам в выравниваниях.
Построение выравнивания по совмещению структур
Выбранные белки:
5A8K - Methyl-coenzyme M reductase from Methanothermobacter wolfeii
1E6Y - Methyl-coenzyme M reductase from Methanosarcina barkeri
5N1Q - Methyl-coenzyme M reductase III from Methanothermococcus thermolithotrophicus
Выравнивание по совмещению структур было построено с помощью сайта PDB (см. рис. 1-3).
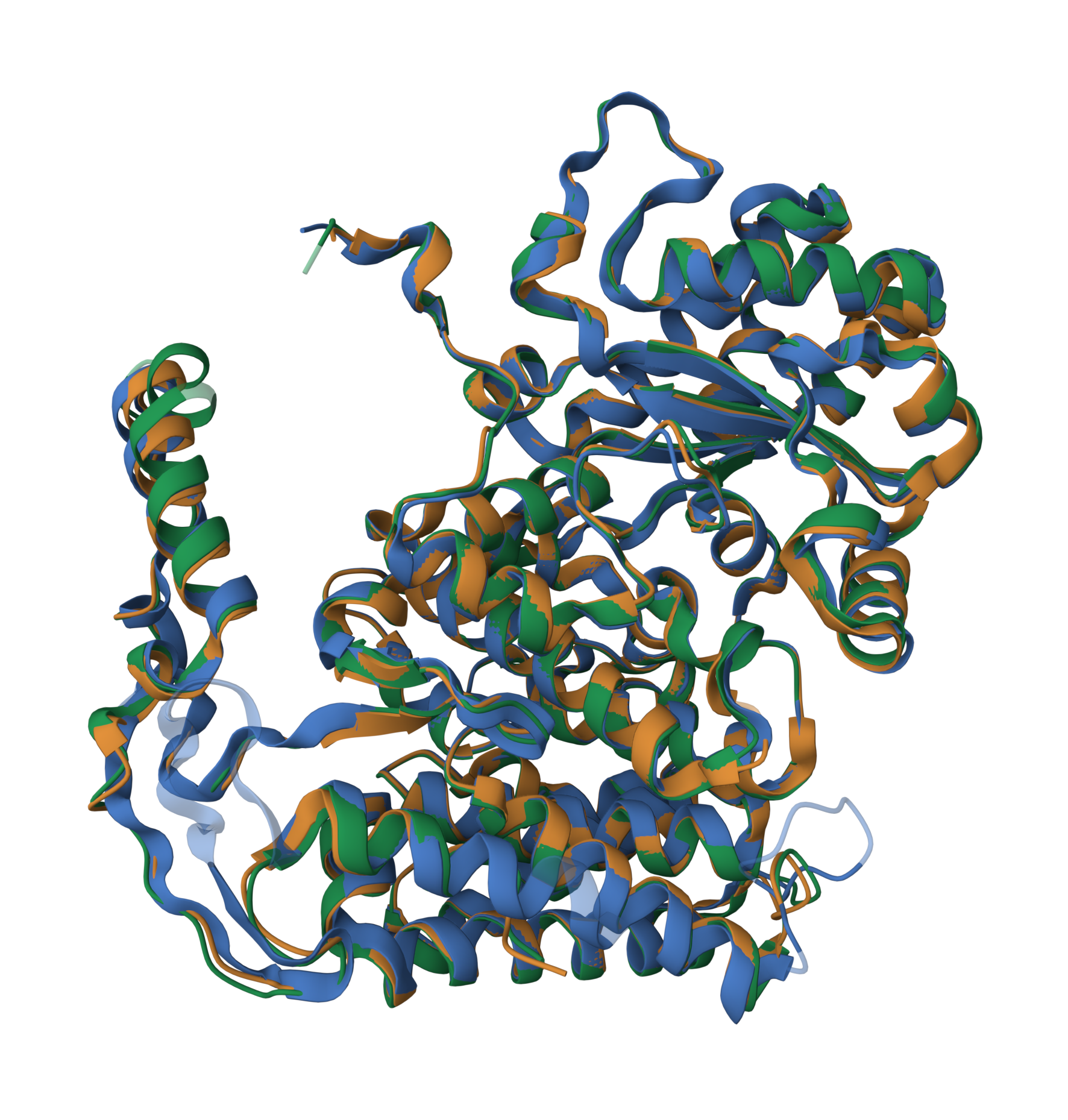
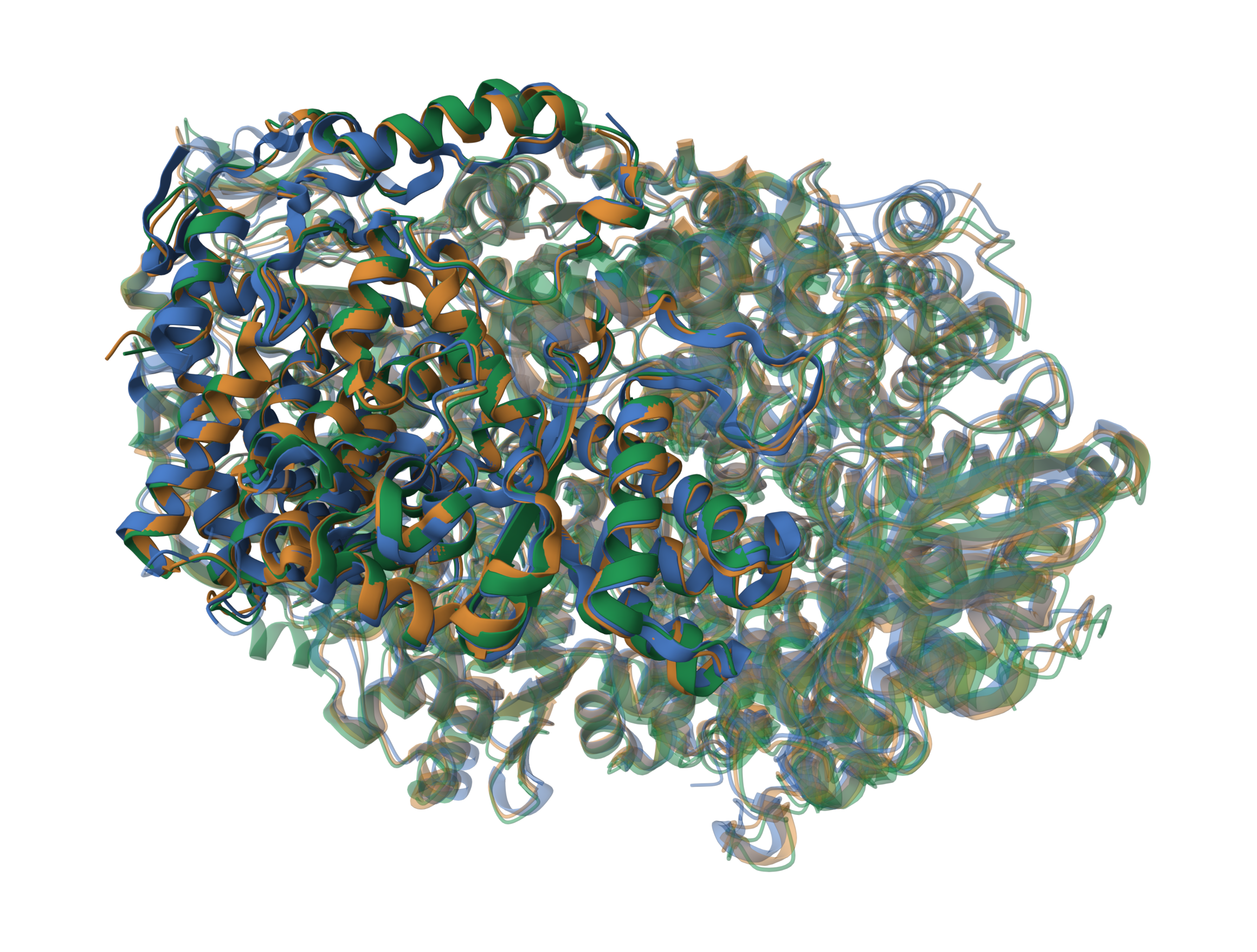
Выравнивание программой Mafft: Все 3 белка имеют по 3 цепи, поэтому выравнивание всех 9 последовательностей непоказательно. Я сделала выравнивание цепей A, чтобы сравнить его с выравниванием PDB. Выравнивание цепей A трех белков: Chain A
Результаты выравниваний: Последовательности очень похожи, много абсолютно идентичных блоков (116-122, 141-147, 337-358, 472-479, 494-511 и др.). Выделяются участки с гэпами ( 21-30, 378-384). Вероятно, белки гомологичны. Также мы видим, что в обоих выравниваниях белки больше всего отличаются у N-конца. Сравнивая разные способы построения выравниваний, можно заметить, что в выравнивании PDB (рис.2) лучше видны невыровненные участки (в основном это неупорядоченные петли).Описание T-Coffee
T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation) обеспечивает значительное повышение точности при незначительных затратах на скорость. Алгоритм имеет две основные особенности. Во-первых, он предоставляет простой способ создания множественных выравниваний с использованием разнородных источников данных (различных библиотек парных выравниваний). Второй важной особенностью T-Coffee является метод оптимизации, который используется для поиска оптимального множественного выравнивания, которое наилучшим образом соответствует парным выравниваниям во входной библиотеке. Следовательно, используется информация из библиотеки для выполнения последовательного выравнивания таким образом, чтобы учитывать выравнивания между всеми парами при выполнении каждого шага последовательного множественного выравнивания.[1] В сравнении с другими алгоритмами MSA, T-Coffee можно назвать достаточно точным, но более медленным. [2]
Литература
1. Notredame C., Higgins D. G., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment //Journal of molecular biology. – 2000. – Т. 302. – №. 1. – С. 205-217. 2. Di Tommaso P. et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension //Nucleic acids research. – 2011. – Т. 39. – №. suppl_2. – С. W13-W17.