Описание полиморфизмов у пациента.
Часть I: подготовка чтений
Я работала в директории /nfs/srv/databases/ngs/ksenia с файлами chr12.fastq и chr12.fasta.
Анализ качества чтений. С помощью программы FasQС было проанализировано качество чтений. Она установлена на kodomo, поэтому я вызвала её командой:
fastqc chr12.fastq
Программа выдала архив файлов chr12_fastqc.zip и отчет о работе программы chr12_fastqc.html.
Очистка чтений. Для очистки чтений использовалась программа Trimmomatic. Необходимо отрезать с конца каждого чтения нуклеотиды с качеством ниже 20 и оставить только чтения длиной не меньше 50 нуклеотидов. Для этого были выбраны параметры TRAILING:20 и MINLEN:50.
TRAILING удаляет с конца чтения нуклеотиды с качеством ниже указанного. MINLEN удаляет чтения с длиной меньше указанного числа.
Использовалась команда (программа Trimmomatic установлена на kodomo): java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr12.fastq chr12_out.fastq TRAILING:20 MINLEN:50
В результате работы программы был получен файл chr12_out.fastq.
Затем я сделала анализ качества очищенных чтений с помощью FastQC. Для этого я использовала команду:
fastqc chr12_out.fastq
В результате работы программы был получен архив файлов chr12_out_fastqc.zip и файл с отчётом chr12_out_fastqc.html.
Сравнение качества до и после чистки
До очистки: Число чтений 7157. Длина чтений 31-100. %GC 37
После очистки: Число чтений 7023. Длина чтений 50-100. %GC 36
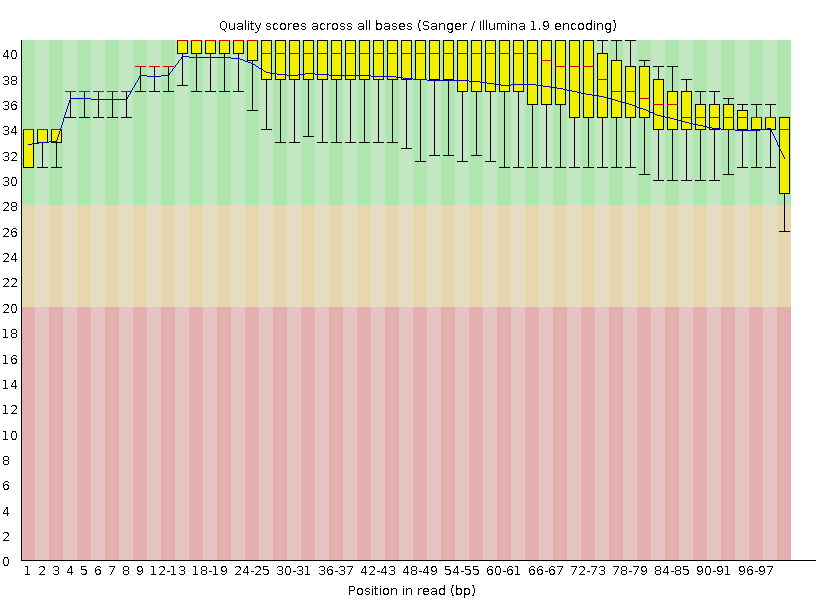
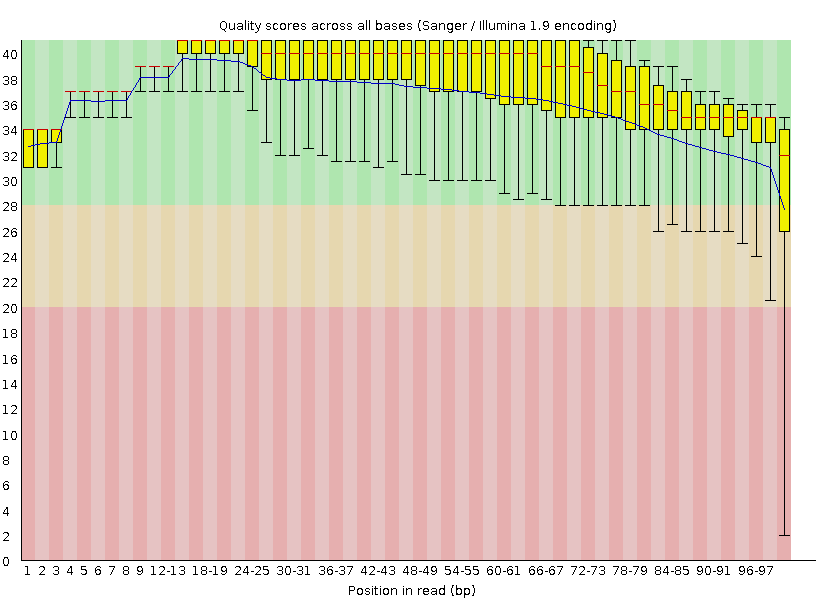
Per base sequence quality для последовательности приемлимо и до чистки и после (программа не выдает предупреждение). Также видно, что после чистки больше нет "усов" (края статистически значимой выборки) выходящих из зеленой области, некоторые интерквартильные рахмахи (желтые прямоугольники) уменьшились, математическое ожидание по качеству (синяя линия) повысилось. То есть качество чтений увеличилось. Таким образом, были вырезаны только низкокачественные чтения и остались чтения пригодные для работы.
Часть II: картирование чтений
Картирование чтений. Очищеные чтения были откартированы с помощью программы BWA, установленнной на kodomo.
Сначала файл chr12.fasta был проиндексирован командой: bwa index chr12.fasta. Получен индексированный файл chr12.fasta.
Затем было построено выравнивание прочтений и референса в формате .sam. Использовалась команда: bwa mem chr12.fasta chr12_out.fastq > chr12.sam. Получен файл chr12.sam.
Анализ выравнивания.
Для начала выравнивание чтений с референсом было переведено в бинарный формат .bam. Используя программу samtools. Использовалась команда: samtools view chr12.sam -b -o chr12.bam. Параметр -b меняет формат выходного файла с установленного по умолчанию, -o обозначат имя выходного файла. Получен файл: chr12.bam.
Далее было отсортировано выравнивание чтений с референсом по координате в референсе начала чтения. Использовалась команда: samtools sort chr12.bam -T smth.txt -o chr12_sort.bam. Параметр -T позволяет записывать временные файлы в файл smth.txt, а не в stdout. Получен файл: chr12_sort.bam
Затем полученный файл был проиндескирован командой: samtools index chr12_sort.bam. Получен проиндексированный файл chr12_sort.bam
Также было выяснено, сколько чтений откартировалось на геном командой: samtools idxstats chr12_sort.bam > chr12.out. Получен файл: chr12.out.
Из файла chr12.out было получено, что на хромосому было откартировано 7024 чтения и 2 чтения не были откартированы.
Часть III: Анализ SNP
Поиск SNP и инделей.
Сначала был создан файл с полиморфизмами в формате .bcf. Команда: samtools mpileup -uf chr12.fasta chr12_sort.bam -o snp.bcf. Параметр -o обозначат имя выходного файла. Получен файл snp.bcf.
Затем был создан файл со списком отличий между референсом и чтениями в формате .vcf. Команда: bcftools call -cv snp.bcf -o snp.vcf. Получен файл snp.vcf
В файле snp.vcf 38 полиморфизма из них 35 замен и 3 делеции. Среднее качество 82.79, среднее покрытие 11.125. Самое лучшее качество 225, самое худшее 3,5427. Самое лучшее покрытие 133, худшее 1.
| Координата | Тип полиморфизма | Качество чтений | Глубина покрытия | В референсе | В чтении |
| 9847808 | Замена | 73.0074 | 12 | A | G |
| 9833524 | Замена | 225.009 | 110 | C | G |
| 9849138 | Делеция | 179.468 | 21 | ataaagtaaa | ataaa |
Аннотация SNP.
С помощью программы ANNOVAR были проаннотированны полученные SNP. Для начала с помощью скрипта convert2annovar.pl нужно было создать файл (без инделей), с которым может работать эта программа. Из файла snp.vcf вручную были удалены индели, получен файл: snp_no.vcf.
Команда для создания нужного файла: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp_no.vcf -outfile snp.avinput
Получен файл: snp.avinput
Для аннотации snp по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar использовался скрипт annotate_variation.pl.
Аннотация по базе refgene.
В результате полученны файлы: chr12.refgene.variant_function; chr12.refgene.exonic_variant_function ; chr12.refgene.log. Файл chr12.refgene.variant_function содержит описание всех полиморфизмов, в первой колонке содержатся категории snp, которые указывают на то где находится полиморфизм (exonic-внутри экзона, intronic-внутри интрона и т. д.). А также в файле указано гетерозиготный (11) или гомозиготный (24) полиморфизм.
| exonic | intronic | UTR3 | ncRNA_intronic | Всего |
| 3 | 25 | 6 | 1 | 35 |
| Координата | Ген | Замена нуклеотидная | Тип замены | Качество чтений | Глубина покрытия | Аминокислотная замена |
| 9822387 | CLEC2D | C > G | несинонимичная, гетерозиготная | 190.000 | 31 | N19K |
| 9833524 | CLEC2D | C > G | несинонимичная, гетерозиготная | 225.009 | 110 | L23V |
| 9833628 | CLEC2D | C > T | синонимичная, гомозиготная | 221.999 | 71 | S57S |
Качество чтений и глубина покрытия в основном низкие. Больше всего полиморфизмов категории intronic, потому что замены в кодирующих последовательностях могут привести к нарушению функции белка и подвергаются более тщательному отбору, чем некодирующие. Качество и грубина покрытия всех трех полиморфизмов категории exonic очень хорошие.
Аннотация по базе DBSNP
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr12.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы: chr17.dbsnp.hg19_snp138_dropped (полиморфизмы имеющие rs); chr12.dbsnp.hg19_snp138_filtered (полиморфизмы не имеющие rs); chr12.dbsnp.log.
Получаем, что 28 полиморфизмов имеют rs (аннотацию в базе данных dbsnp), 7 не имеют. При этом все не аннонтированные полиморфизмы имеют плохое качество и покрытие, а в аннонтированных есть и хорошее качество и покрытие (хотя плохого много).
Аннотация по базе 1000 genomes
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr12.1000 -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы: chr12.1000.hg19_ALL.sites.2014_10_dropped ; chr12.1000.hg19_ALL.sites.2014_10_filtered; chr12.1000.log.
Получаем, что 28 полиморфизмов аннонтировано, а 7 нет. То есть результат такой же, как при аннотации по базе данных dbsnp. По качеству и покрытию ситуация такая же, только в этой аннотации есть еще один параметр - частота. Она для аннонтированных варьируется от 0.966254 до 0.0107827 (для неаннонтированных такого параметра нет).
Аннотация по базе GWAS
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out chr12.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы: chr12.gwas.hg19_gwasCatalog (содержит snp, которые имеют клиническое значение); chr12.gwas.log.
Аннотация содержит только 4 snp с известным клиническим значением.
| Координата | Значение |
| 9833628 | Type 1 diabetes | Диабет 1-го типа |
| 66343810 | Brain structure | Структура мозга |
| 66359752 | Height | Рост |
| 107367225 | Bone mineral density | Минеральная плотность костной ткани |
Аннотация по базе Clinvar
Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr12.clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/
Полученные файлы: chr12.clinvar.hg19_clinvar_20150629_dropped (содержит аннотированные snp); chr12.clinvar.hg19_clinvar_20150629_filtered; chr12.clinvar.log.
Получаем, что не аннонтированн ни один snp.
Все найденые snp и их характеристики по базам данных были собраны в сводную таблицу snp.xlsx. Использовались только аннонсированные (dropped файлы) snp.
Ссылки:
© Кузнецова Ксения, 2015