Поиск сигналов. chip-seq
Мне был дан файл chipseq_y14/chipseq_chunk10.fastq в котором риды Illumina, полученные в результате сhip-seq эксперимента, разделены на отдельные файлы, соответствующие участкам хромосом.
Контроль качества чтений.
С помощью программы FastQC был проведен контроль качества чтений.
Команда: fastqc chipseq_chunk10.fastq
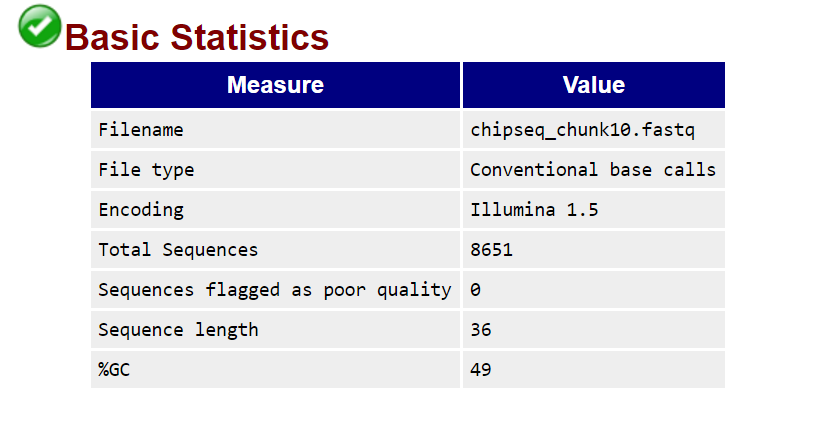
Программа выдала 2 файла: chipseq_chunk10_fastqc.html и chipseq_chunk10_fastqc.zip.
На рисунке ниже приведена информация о чтениях.
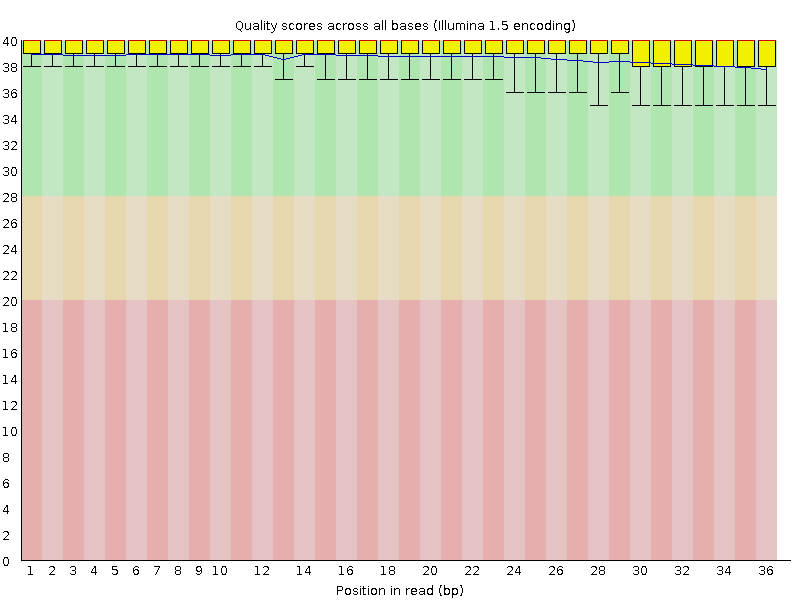
На рисунке ниже представлено качество чтений. Все риды отмечены как хорошие. Даже усы не выходят из зеленой зоны. Поэтому очищать чтения с помощью Trimmomatic нет необходимости.
Картирование прочтений.
Затем было проведено картирование чтений на геном человека hg19 программаой BWA. Предоставленный геном уже проиндексирован, следовательно необходимости в индексировании нет.
Команда: bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk10.fastq > chipseq_chunk10.sam
Был получен файл chipseq_chunk10.sam и проведен его анализ.
samtools view -b chipseq_chunk10.sam -o chunk10.bam Эта команда переводит в бинарный формат.
samtools sort chipseq_chunk10.bam -T chip_temp -o chipseq_chunk10.sorted.bam Эта команда сортирует по координате в референсе начала чтений.
samtools index chunk10_sorted.bam Эта команда индексирует отсортированное.
samtools idxstats chunk10_sorted.bam Эта команда выдает информацию о количестве чтений, откартированных на геном.
samtools view -c chipseq_chunk10.sorted.bam Эта команда выводит на экран общее количество откартированных чтений.
Изначально было 8651 рида. Стало 8651. На геном откартированно столько ридов сколько и было. На хромосому 1 откартировалось большинство чтений, следовательно можно предположить, что мне для анализа были предложены прочтения с 1й хромосомы.
Поиск пиков.
С помощью программы MACS, установленной на кодомо, был выполнен поиск пиков.
macs2 callpeak -t chipseq_chunk10.sorted.bam --nomodel -n Chunk
Получены 3 файла: Chunk_peaks.narrowPeak , Chunk_peaks.xls, Chunk_summits.bed.
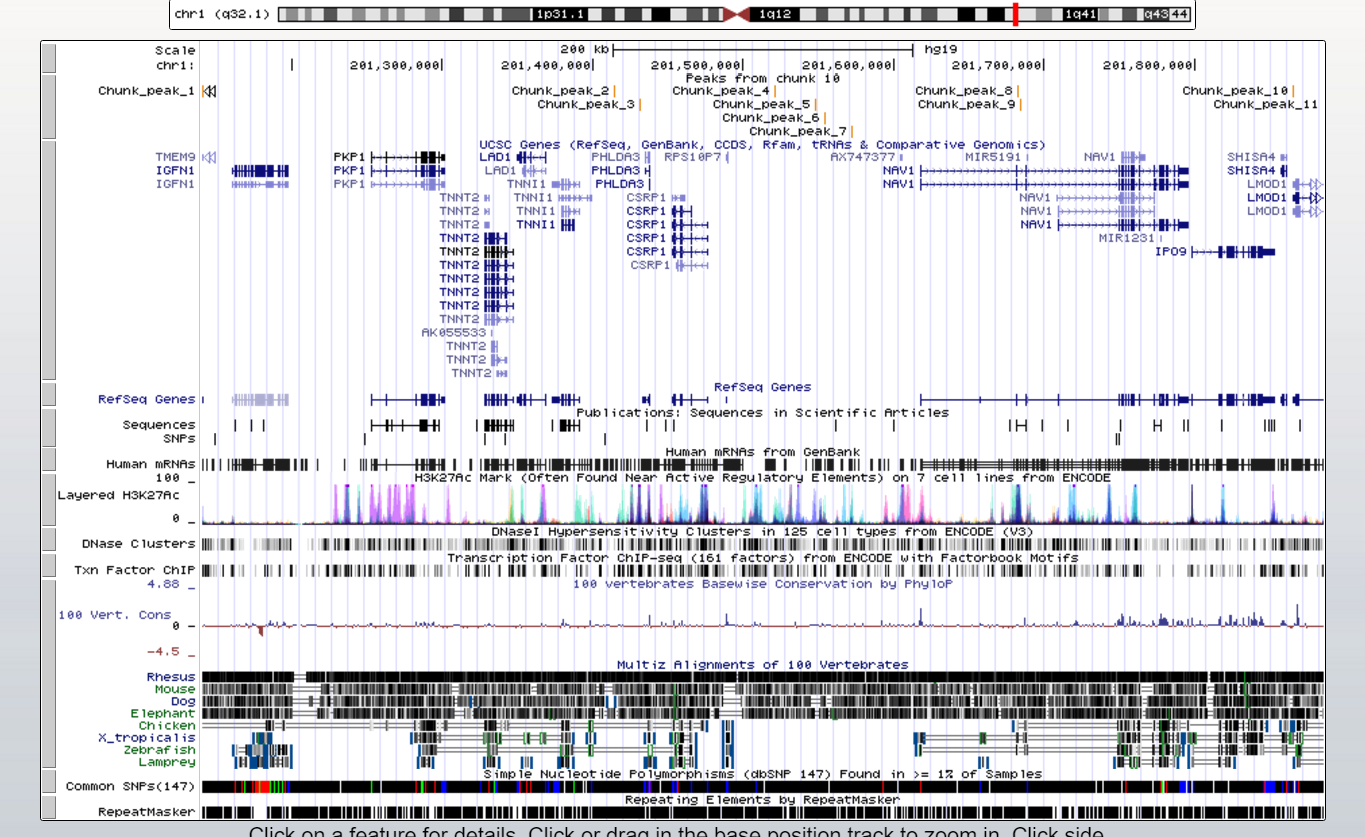
Всего найдено 11 пиков. Все они в 1й хромосоме. Ширина пиков от 217 до 392.
| name | start | end | length | abs_summit | pileup | -log10(pvalue) | fold_enrichment | -log10(qvalue) |
| Chunk_peak_1 | 201140633 | 201140896 | 264 | 201140774 | 34.00 | 22.60547 | 7.84753 | 16.05741 |
| Chunk_peak_2 | 201413996 | 201414237 | 242 | 201414129 | 31.00 | 28.15574 | 11.51079 | 21.39019 |
| Chunk_peak_3 | 201431086 | 201431365 | 280 | 201431241 | 40.00 | 33.87111 | 11.51685 | 26.80800 |
| Chunk_peak_4 | 201520986 | 201521254 | 269 | 201521119 | 43.00 | 33.66709 | 10.52632 | 26.61922 |
| Chunk_peak_5 | 201547477 | 201547796 | 320 | 201547568 | 24.00 | 13.66005 | 5.84112 | 7.40290 |
| Chunk_peak_6 | 201554247 | 201554463 | 217 | 201554331 | 25.00 | 16.98054 | 7.26257 | 10.60839 |
| Chunk_peak_7 | 201572049 | 201572248 | 200 | 201572128 | 20.00 | 12.71633 | 6.17647 | 6.50442 |
| Chunk_peak_8 | 201682607 | 201682828 | 222 | 201682736 | 38.00 | 27.48340 | 9.06977 | 20.77094 |
| Chunk_peak_9 | 201683801 | 201684081 | 281 | 201683954 | 44.00 | 34.36695 | 10.56338 | 27.28265 |
| Chunk_peak_10 | 201865056 | 201865447 | 392 | 201865261 | 97.00 | 100.32639 | 21.03004 | 90.86187 |
| Chunk_peak_11 | 201885843 | 201886061 | 219 | 201885947 | 25.00 | 19.40718 | 8.55263 | 12.96592 |
Самый достоверный пик с наименьшим p-value, так как у нас -log10(pvalue), то достовернее тот у которого этот параметр выше. А именно пик 10, а наимее пик 7.
Затем информация из файла Chunk_peaks.narrowPeak была визуализирована с помощью UCSC Genome Browser. Для этого в начало файла были добавлены строчки:
track type=narrowPeak visibility=3 db=hg19 name="Chunk_peaks" description="Peaks from chunk 10"
browser position chr1:201140642-201886071
Рассмотрим 2 наиболее достоверных пика 10 и 9. Также они из самых длинных пиков. Расстояние от начала до вершины пика 9 = 153, пика 10 = 205. Обе вершины находятся ближе к концу пика.
Ниже на рисунках представлены увеличенные изображения пиков 9 и 10.
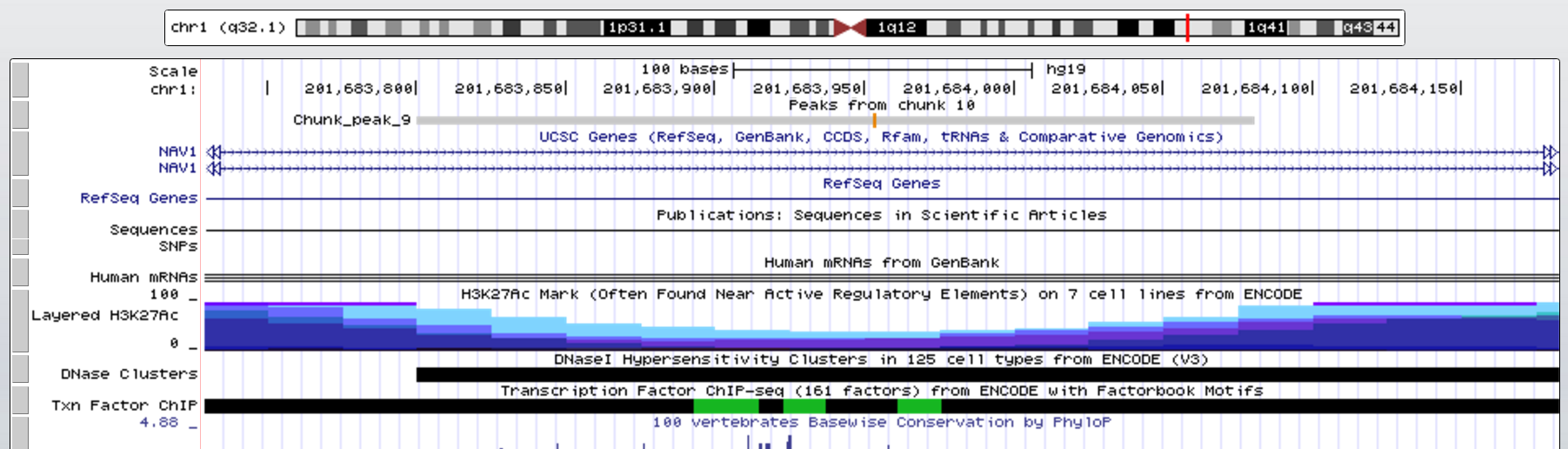
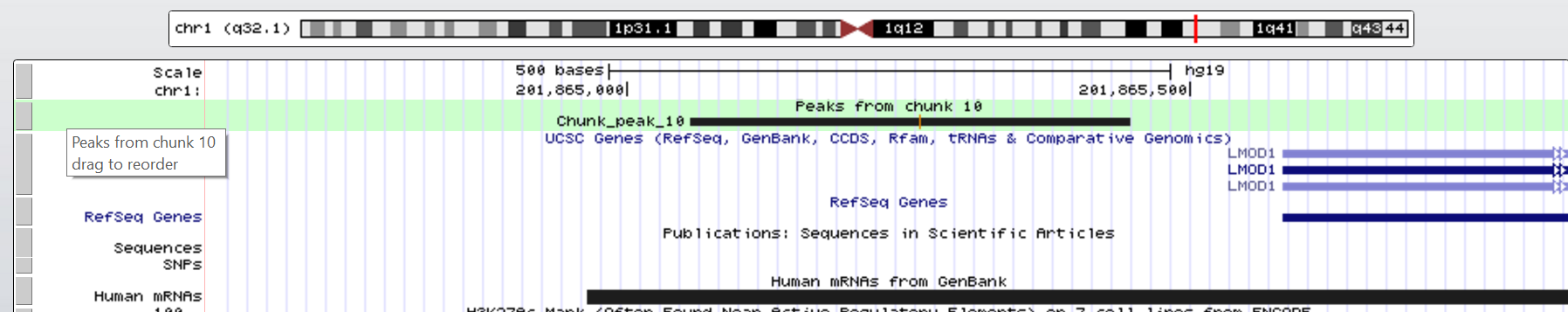
Пик 9 перекрввается с NAV1_HUMAN (neuron navigator 1), который кодирует белок связанный с различной клеточной деятельностью, экспрессируются в нервную систему. Пик 10 расположен перед геном LMOD1_HUMAN (leiomodin 1), кодирует белки гладкой мускулатуры.
Ссылки:
© Кузнецова Ксения, 2015