|
||||
|
|
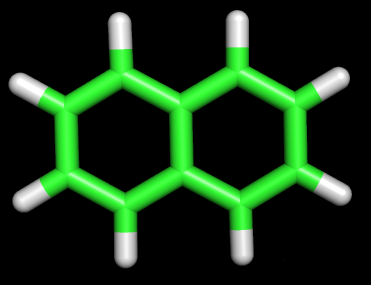
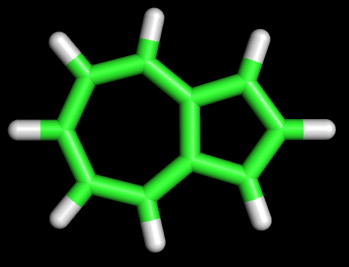
1 C помощью MOPAC были оптимизированы структуры нафталена и азулена:
Была проведена оптимизация геометрии для обеих молекул с помощью GAMESS: gms naph_opt.inp 1 >& naph_opt.log gms azu_opt.inp 1 >& azu_opt.logПолученные файлы naph_opt.log и azu_opt.log 3 На основе полученных координат были составлены новые входные файлы для расчёта энергии. Были построены по два файла на каждую молекулу, в первом случае для расчёта методом Хартри-Фока, во втором - с помощью теории функционала плотности. Для расчёта по Хартри-Фоку файл с заголовком $CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATAДля рассчета с помощью теории функционала плотности заголовок файла $CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATAПолученные файлы для рассчета: naph_HF.inp, naph_func.inp, azu_HF.inp, azu_func.inp. Были рассчитаны четыре системы: два способа на каждую молекулу. 4 Из log файлов расчёта энергии из строки "TOTAL ENERGY = " были выписаны значения энергии в таблицу:
|
© Яшина 2009