|
||||
|
Семи-эмпирические вычисления: MOPACНачнём работу с установки переменных:
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Задание 1На основе аннотации SMILES порфирина был создан файл 1.smi. C помощью obgen создадим файл с 3D структурой порфирина и откроем ее в PyMOL.Команда: obgen 1.smi > 1.molПолученный файл 1.mol. Изображение: 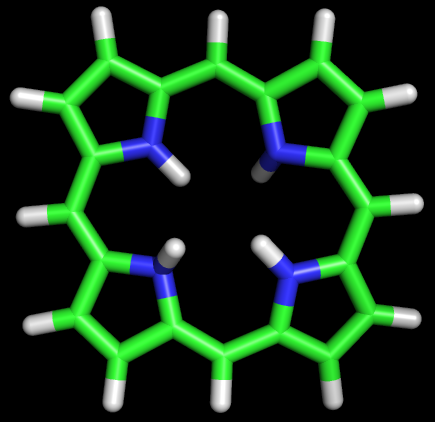 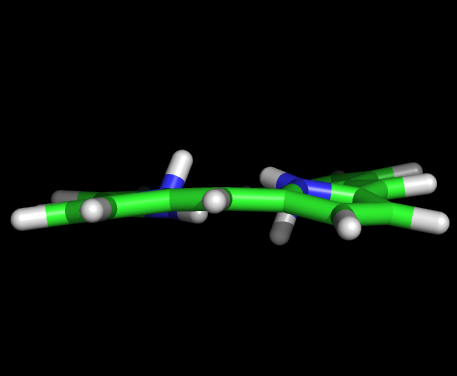 В структуре удалили 2 водорода, нарушавших ароматичность, и сохранили файл в формате pdb (1.pdb). С помощью babel переформатировали координаты во входной файл для Mopac babel -ipdb myfile.pdb -omop 1_opt.mop -xk "PM6"Полученный файл 1_opt.mop. Затем запустили Mopac: MOPAC2009.exe 1_opt.mopФайл вывода out 1_opt.out переформатировали в pdb: babel -imopout 1_opt.out -opdb 1_opt.pdbПосле открытия полученого pdb файла в PyMOL обнаружилось, что молекула порфирина стала плоской, что согласуется с восстановлением ароматичности после удаления двух водородов. Изображение: 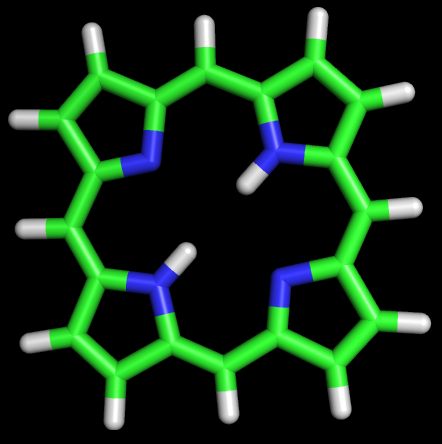  Задание 2Было необходимо рассчитать возбужденные состояния порфирина и на основе этих данных оценить спектр поглощения молекулы. Для расчёта возбуждённых состояний была сделана копия mop файла из предыдущего задания 1_opt_spectr.mop. Для указания Mopac о необходимости расчёта возбуждённого состояния в конец файла были добавлены строки:
пустая строка
cis c.i.=4 meci oldgeo
some description
Полученный файл 1_opt_spectr.mop. Затем был запущен Mopac:MOPAC2009.exe 1_opt_spectr.mopПолученный файл 1_opt_spectr.out. Значения энергий для электронных переходов в конце файла были пересчитаны в длину волн, при которых происходят эти переходы: ENERGY(EV) WAVE LENGTH(nm) 1.914365 648 2.266802 547 2.464170 503 2.825733 439 3.364386 369 3.391543 366 3.669162 338 3.871959 320 Задание 3Для молекулы O=C1C=CC(=O)C=C1 была определена геометрия с помощью obgen и с помощью Мopac (см. задание 1).Файл 3.smi был подан на вход программе obgen, получен файл 3.mol. C помощью babel он был преобразован во входной для MOPAC формат (файл 3_opt.mop). После запуска MOPAC был получен файл 3_opt.out, который был преобразован в формат pdb (3_opt.pdb). Изображение полученных структур, наложенных друг на друга, зеленым цветом представлены атомы углерода из 3_opt.pdb, голубым - из 3.pdb: 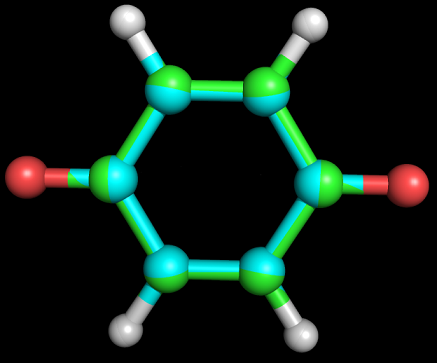 Затем был преобразован файл 3_opt.mop: были добалена строка CHARGE=-2, а также явным способом указаны атомы, накоторых должен находиться отрицательный заряд. Полученный файл 3_opt_dianion.mop. С помощью MOPAC был получен файл 3_opt_dianion.out, который затем был преобразован в pdb файл 3_opt_dianion.pdb. Изображения структур 3_opt.pdb и 3_opt_dianion.pdb, наложенные друг на друга, атомы углерода для первой структуры зеленые, для второй - красные: 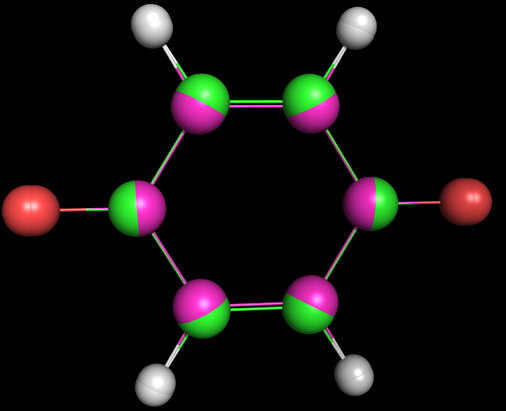 Заметно, что молекула дианиона вытянута вдоль оси, соединяющей кислороды, что можно объяснить взаимным отталкиванием двух отрицательно заряженных атомов. |
© Яшина 2009