| Команда | Описание |
|---|---|
| hisat2-build chr11.fasta indexed | Индексирует референсную последовательность. |
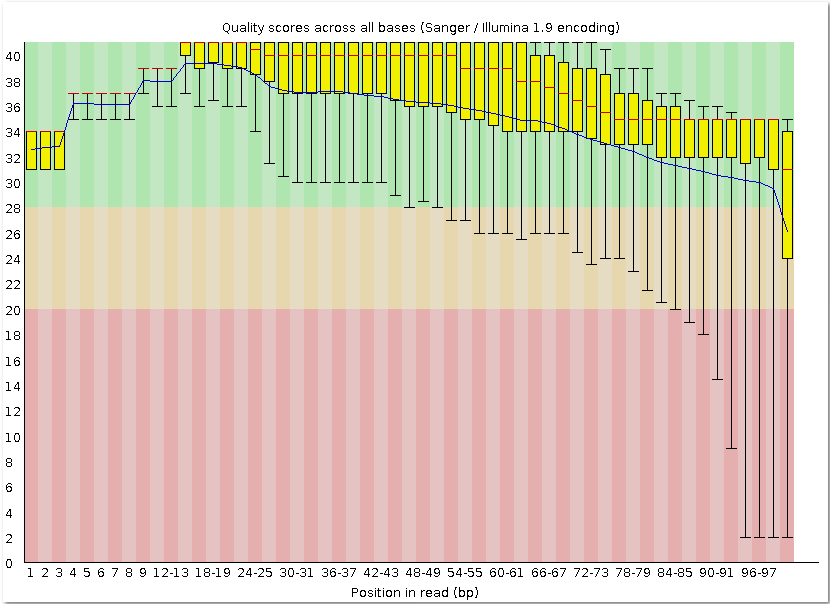
| fastqc chr11.fastq | Контроль качества чтения chr11.fastq с помощью программы FastQC. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr11.fastq outfile.fastq TRAILING:20 MINLEN:50 |
Программа Trimmomatic очищает чтения. В данном задании требовалось убрать с каждого конца чтения нуклеотиды с качеством ниже 20 и оставить чтения длиной не меньше 50 нуклеотидов. |
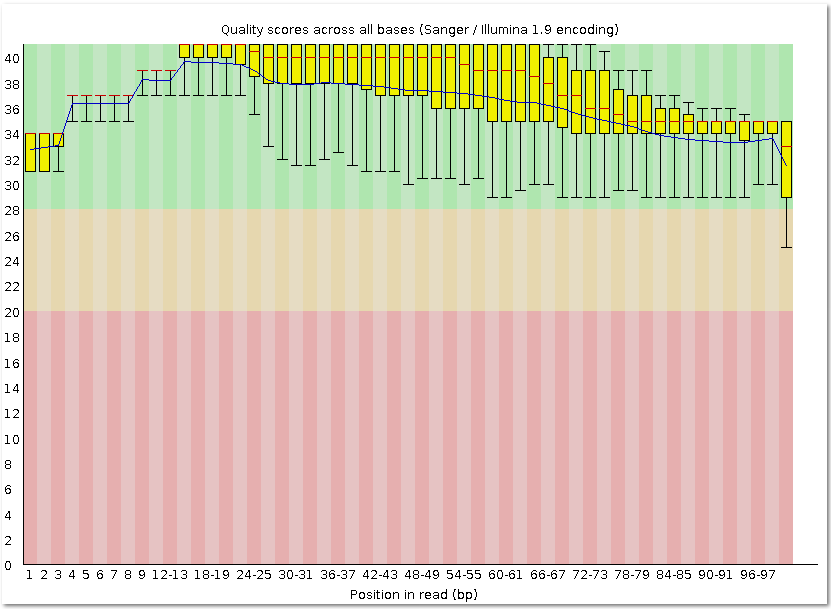
| fastqc outfile.fastq | Контроль качества чтений, прошедших триммирование. |
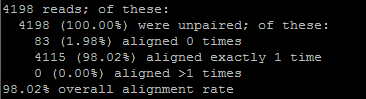
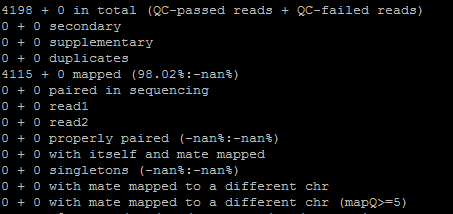
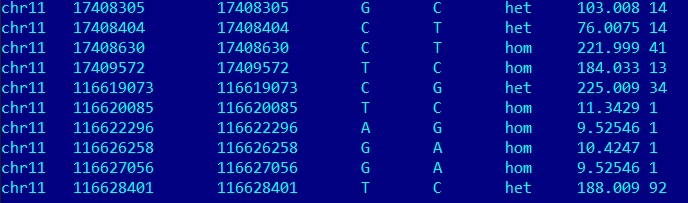
| POS-координата | REF-референс | ALT-риды | INFO-тип | DP-глубина покрытия | QUAL-качество ридов |
|---|---|---|---|---|---|
| 17408305 | G | C | SNP(трансверсия) | 14 | 103.008 |
| 116620085 | T | C | SNP(транзиция) | 1 | 11.3429 |
| 116628401 | T | C | SNP(транзиция) | 92 | 188.009 |
| Команда | Вывод | Пояснение |
|---|---|---|
| annotate_variation.pl -filter -out dbnsp.snp -build hg19 -dbtype snp138 chr11.avinput /nfs/srv/databases/annovar/humandb.old/ |
1.dbnsp.snp.hg19_snp138_dropped 2.dbnsp.snp.hg19_snp138_filtered 3.dbnsp.snp.log |
1. В файле те замены, которые содержат rs в базе snp138, то есть имеют идентификатор в SNP. (9) 2. В файле замены, которых нет в базе snp138 (не имеют идентификатора rs). (1) 3. Комментарии к работе программы. |
| annotate_variation.pl -filter -out 1000genomes -build hg19 -dbtype 1000g2014oct_all chr11.avinput /nfs/srv/databases/annovar/humandb.old/ | 1. 1000genomes.hg19_ALL.sites.2014_10_dropped - содержит полиморфизмы, имеющие rs в 1000 genomes, и их частоты. 2. 1000genomes.hg19_ALL.sites.2014_10_filtered -содержит полиморфизмы, не имеющие rs в 1000 genomes. 3. 1000genomes.log - содержит отчет о работе команды. |
1. Cодержит замены, имеющие rs в базе данных 2014 года 1000 genomes, и столбец с их частототами.(9) 2. Cодержит замены, не имеющие rs в 1000 genomes. (1) 3. Содержит отчет о работе команды. |
| annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog chr11.avinput /nfs/srv/databases/annovar/humandb.old/ | 1.gwas.hg19_gwasCatalog 2.gwas.log |
1. Содержится 4 строки с 4мя заменами пациента, которые есть в базе gwas Catalog. В файле записываются только названия ассоциированной с полиморфизмом болезни:
2 diabetes, Triglycerides, Triglycerides-Blood Pressure (TG-BP). 2.Содержит отчет о работе команды. |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out clinvar chr11.avinput /nfs/srv/databases/annovar/humandb.old/ | 1. clinvar.hg19_clinvar_20150629_dropped 2. clinvar.hg19_clinvar_20150629_filtered 3. clinvar.log |
1. Содержит SNP с известным клиническим значением. Permanent neonatal diabetes mellitus (Неонатальный сахарный диабет) 2.Содержит замены, не имеющие rs в Clinvar. 3.Содержит отчет о работе команды. |
| /nfs/srv/databases/ngs/kurkino$ annotate_variation.pl -out refgene -build hg19 chr11.avinput /nfs/srv/databases/annovar/humandb.old/ | 1.refgene.variant_function - содержит описание всех полиморфизмов. 2.refgene.exonic_variant_function - содержит описание полиморфизмов внутри экзонов. 3.refgene.log - содержит отчет о работе команды. |
1.Содержит сами замены с указанием, в каком экзоне\интроне находится замена.
Все мутации разделены на группы: exonic (2), intronic (5), UTR3(3). exonic - полиморфизм внутри экзона (частично или полностью) UTR3 - полиморфизм полностью или частично входит в 3-нетранслируемую область intronic - полиморфизм полностью или частично внутри интрона 2.Содержит только мутации в экзонах, для них указано, являются они синонимическими или нет. В этом файле 2 строки, то есть нашлось 2 замены, лежащие в экзонах. 3.Содержит отчет о работе команды. |