| Команда | Описание | Вывод |
|---|---|---|
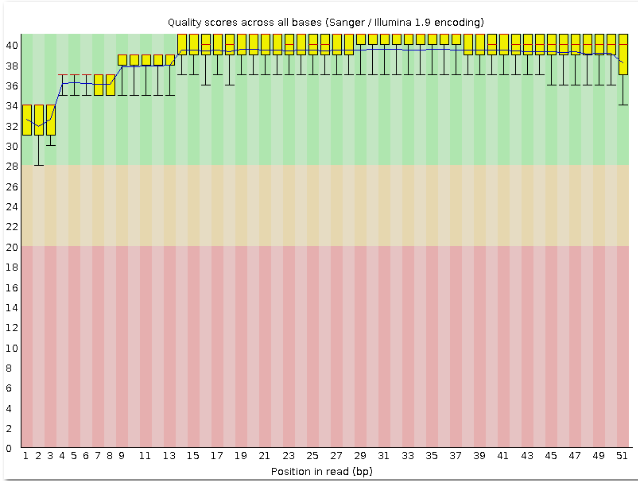
| fastqc chr11.1.fastq | Контроль качества чтения chr11.fastq с помощью программы FastQC. | Были созданы файл chr11.1.fastq и chr11.1_fastqс.zip |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr11.1.fastq outfile.fastq TRAILING:20 MINLEN:50 |
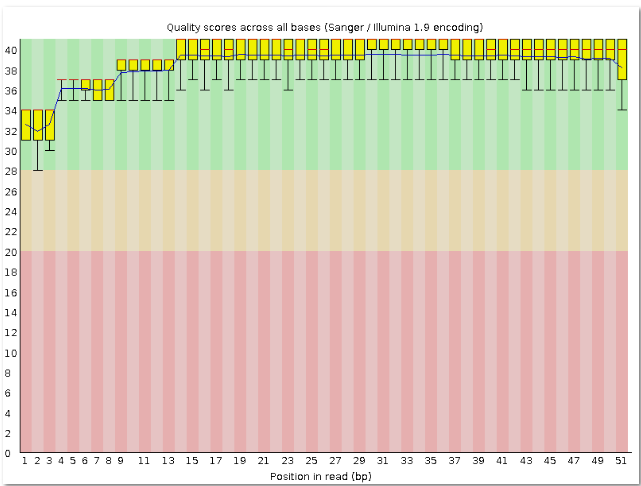
Программа Trimmomatic очищает чтения. В данном задании требовалось убрать с каждого конца чтения нуклеотиды с качеством ниже 20 и оставить чтения длиной не меньше 50 нуклеотидов. | Был создан файл chr11.1_fastq.html.До работы программы было 39549, а осталось 39411 чтений, то есть ушло 0.35% чтений. |
| fastqc outfile.fastq | Контроль качества чтений, прошедших триммирование. | Архив с соответсвующими изображениями. |