 |
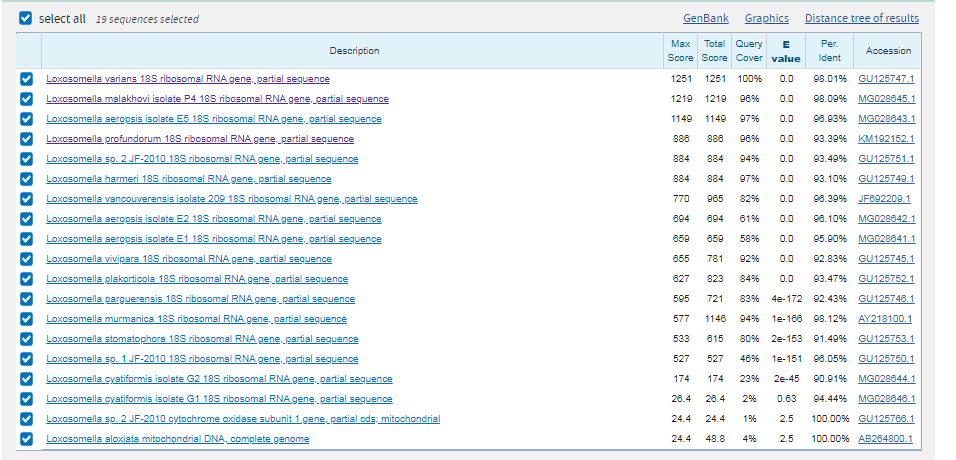
Основной компонент нуклеосомы. Нуклеосомы оборачивают и компактируют ДНК в хроматин, ограничивая доступность ДНК для клеточных механизмов,
которые требуют ДНК в качестве шаблона.
Таким образом, гистоны играют центральную роль в регуляции транскрипции, репарации ДНК, репликации ДНК и хромосомной стабильности.
|
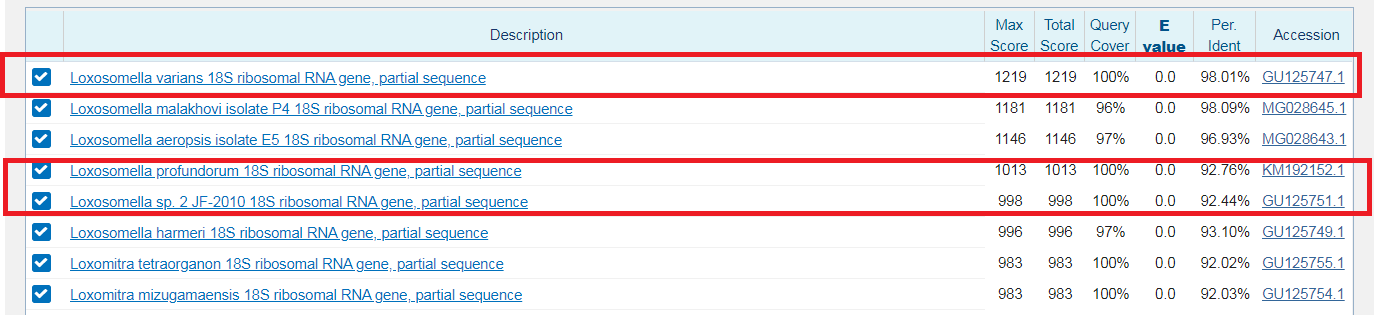
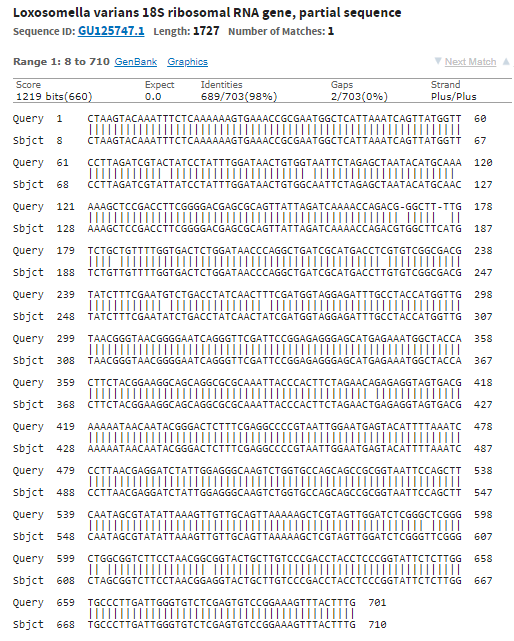
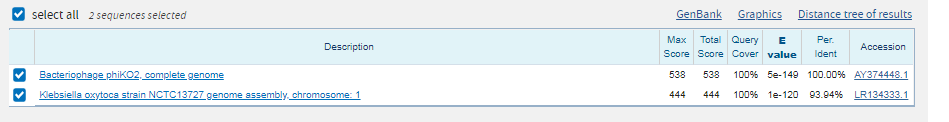
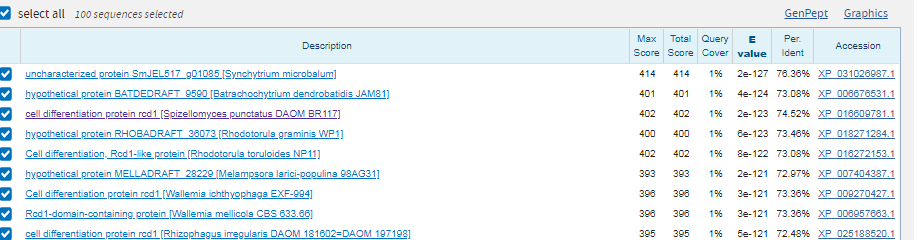
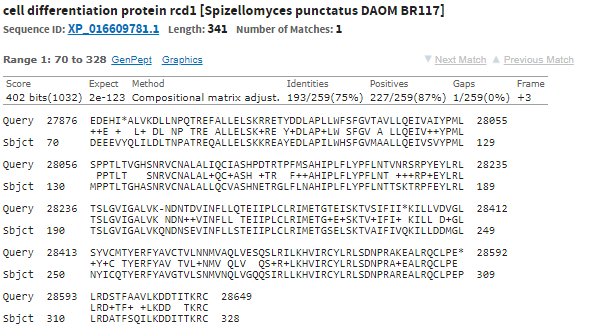
Лучший результат: scaffold-126,вес 244,E-value=2e-74. Покртыие 100%. Все это дает основание полагать, что полседовательности гомологично и функционально схожи.
|
 |
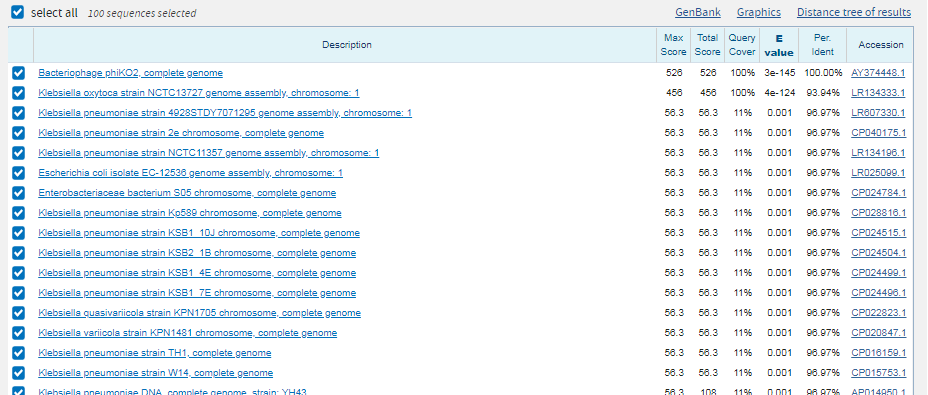
Фермент, катализирующий обратимую реакцию переноса фосфатной группы от 1,3-дифосфоглицериновой кислоты к АДФ,
в результате которой образуются 3-фосфоглицерат и АТФ. |
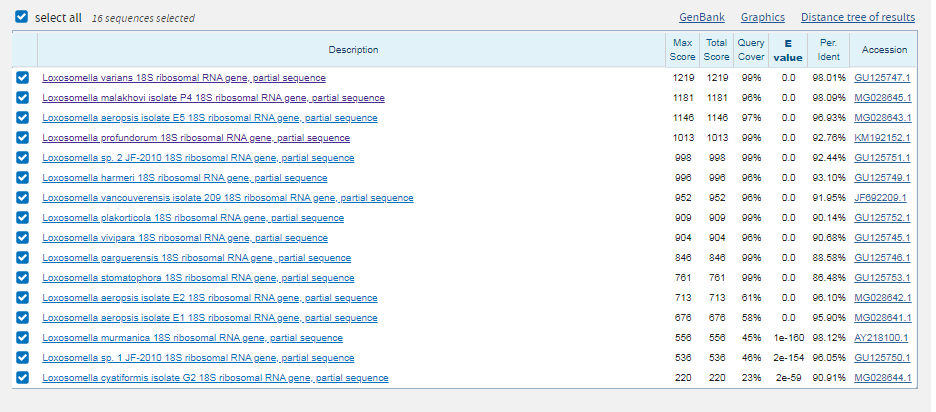
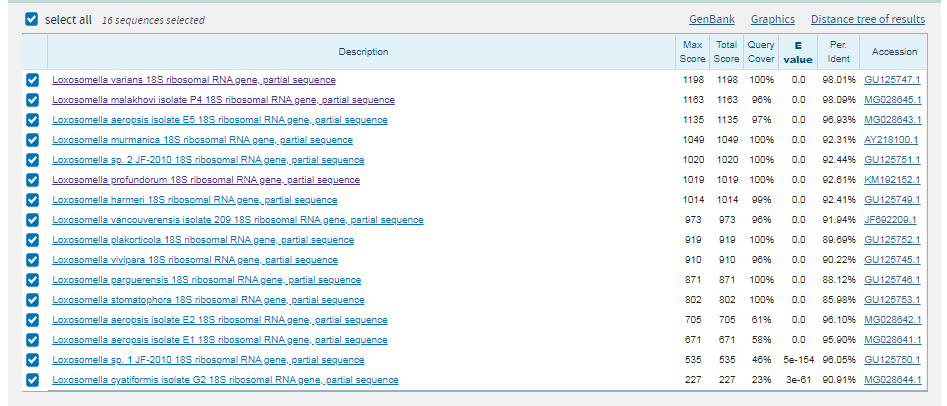
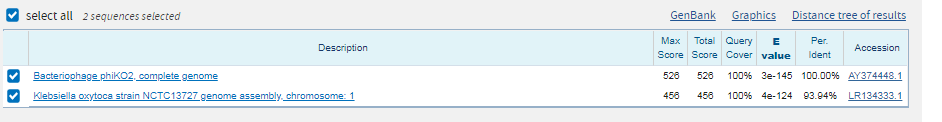
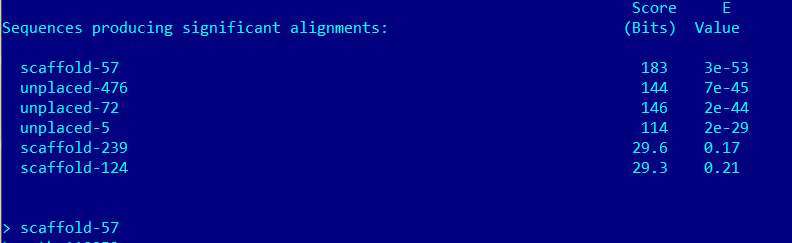
Находка положительная. Покрытие при выравнивании с scaffold-693: вес 484 и E-value=5e-153 составляет 100 %. Опять же есть длинный разрыв, но в остальном последовательность консервативна.
2 последовательности являются гомологами. |