Секвенирование по Сэнгеру
Задание 1. Отчет о работе с последовательностью ДНК на основании данных, полученных из капиллярного секвенатора. Чтение хроматограмм.
Ссылки на файлы
В ходе выполнения задания были проанализированы данные, полученные из капиллярного секвенатора. Исходные файлы можно скачать по ссылкам: прямая последовательность и обратная последовательность. Перед началом работы для оценки количества проблемных нуклеотидов было произведено выравнивание последовательностей с помощью Jalview (программа TCoffee с параметрами по умолчанию), и с этой целью обратная последовательность была преобразована в комплементарную и перевернута. Файлы формата fasta, использованные в ходе работы, можно увидеть по ссылкам: прямая и комплементарная обратной. Выравнивание последовательностей представлено здесь в формате проекта Jalview и fasta-файла. Оценка выравнивания на глаз позволяет сказать, что, несмотря на то, что качество прочтения хроматограмм в начале и в конце оставляет желать лучшего (многие нуклеотиды вообще не распознаны и заменены на N, совпадений мало), в целом процент совпадения очень велик и в средней части последовательности расхождения лишь единичные. Эти расхождения, а также общий уровень шума были рассмотрены с использованием программы UGENE. Сначала обе последовательности были загружены с прямой последовательностью в формате fasta в качестве референса, и был собран консенсус, затем этот консенсус уже был взят в качестве референса и отредактирован. В приложенном ниже файле с проектом UGENE можно увидеть все внесенные изменения и сравнить изначально полученный консенсус с исправленным, где отредактированные нуклеотиды выделены цветом.
Результат работы с UGENE представлен в базе данных UGENE. Отдельно выравнивание двyх последовательностей на консенсус можно посмотреть в файле, импортированном из UGENE, или в проекте Jalview. Также прикрепляю файл с выравниванием на консенсус в формате fasta, fasta-файл с консенсусной последовательностью и fasta-файл с контигом - участком, на котором имеются данные и о прямой, и об обратной цепи. В последнем файле и во всех представленных выравниваниях консенсус и обе последовательности уже отредактированы с помощью UGENE.
Характеристика хроматограммы
Вполне закономерно несколько десятков нуклеотидов с начала и с конца хроматограммы не поддаются точной расшифровке. На глаз я предположила, что достаточно точно можно прочитать последовательность со 106 по 670 нуклеотид, программа же автоматически обрезала до 103 в начале и после 711 в конце. Для обратной последовательности я определила "читаемые" границы на глаз с 70 по 706 нуклеотид, программа же обрезает до 82 в начале и совсем не обрезает конец, расшифровывая все 718 нуклеотидов до конца. В моих оценках имеются расхождения с "решением" программы как в большую, так и в меньшую сторону, что подчеркивает субъективность взгляда одного конкретного исследователя в сравнении с алгоритмом. В связи с плохим качеством хроматограммы было решено не заменять N на букву какого-либо нуклеотида в конечных участках, где нет комплементарной цепи для сравнения пиков.
Оценивая уровень шума визуально, можно отметить в целом высокое качество хроматограммы, отсутствие делеций и вызванных ими "сдвигов" пиков. Спорные моменты, предположительно связанные с гетерозиготностью или мутацией, присутствуют, но единичны. На глаз шум не сливается с сигналом, справа показан фрагмент с наиболее выраженными вторичными пиками. Дефекты ПЦР - пятна красителя - также встречаются, но не препятствуют сборке контига, как как на месте "пятна" в одной цепи вторая прочитана без проблем, и дефект может быть отредактирован.
Для редактирования fasta-файлов последовательностей, а также для оценки уровня шума и качества прочтения были использованы пакеты bio3d и sangerseqR в R. Первый позволяет работать с форматом fasta, второй - с файлами ab1. Уровень шума можно примерно оценить, сравнив высоту первичных и вторичных пиков, хотя, согласно документации, информация о вторичных пиках используется больше для описания полиморфизмов. В R был выполнен следующий код:
library(sangerseqR)
f_seq <- read.abif("C:/Users/user/12_F.ab1")
r_seq <- read.abif("C:/Users/user/12_R.ab1")
mean(f_seq@data$P1AM.1[103:711])
mean(r_seq@data$P1AM.1[82:718])
mean(f_seq@data$P2AM.1[103:711])
mean(r_seq@data$P2AM.1[82:718])
mean(f_seq@data$PCON.1[103:711])
mean(r_seq@data$PCON.1[82:718])
Представленный код считывает две последовательности и далее считает средние значения по нужным полям полученного файла. P1AM.1 и P2AM.1 содержат высоты первичных и вторичных пиков соответственно, PCON.1 - качество прочтения для каждого нуклеотида. С учетом того, что в начале и в конце хроматограммы присутствуют "пятна" красителя и шум неотличим от сигнала, для анализа была выбрана только часть последовательности, вырезанная UGENE как пригодная для анализа. Соответственно, это 103-711 нуклеотиды для прямой последовательности и 82-718 для обратной. На этих отрезках получаем следующее:
| первичный пик | вторичный пик | |
|---|---|---|
| прямая | 1039 (264-2281) | 179 (18-1081) |
| обратная | 1056 (323-2694) | 240 (11-9692) |
Полученные значения в целом свидетельствуют о том, что средняя высота вторичного пика соответсвует минимальной для первичного и лишь единичные из них приближаются по величине к первичным, следовательно, анализ хроматограммы не должен вызвать значительных затруднений. Однако мы можем наблюдать аномально высокие значения высоты вторичного пика для обратной последовательности, локализованные ближе к её началу и объяснимые этим фактом. Изображение фрагмента с аномально высокими вторичными пиками представлено справа. Вероятнее всего, их появление вызвано ошибкой ПЦР или "пятном" красителя, так как их ширина не соответвует типичной для пика, и в прямой последовательности аденин и гуанин в данном районе отсутствуют. Консенсус был отредактирован по прямой последовательности.
Помимо высоты пиков, sangerseqR позволяет охарактеризовать качество прочтения. Этот параметр содержится в поле PCON.1, и для прямой последовательности его среднее значение составляет 29, для обратной - 31. Хроматограмма считается достаточно качественной, если этот параметр выше 20, очень хорошей - если выше 30.
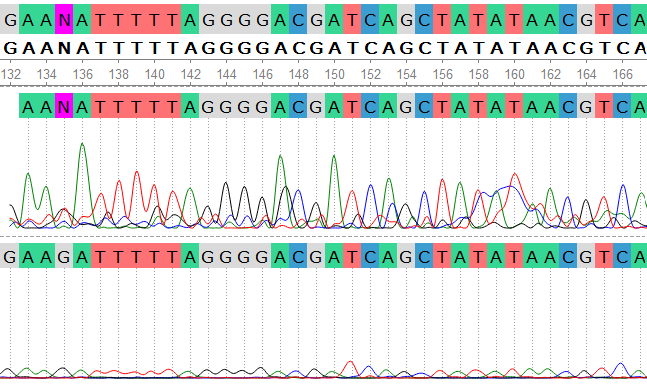
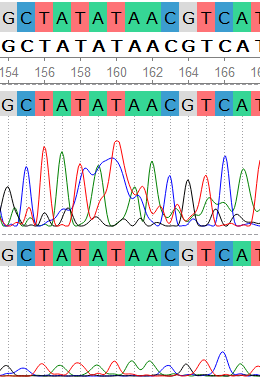
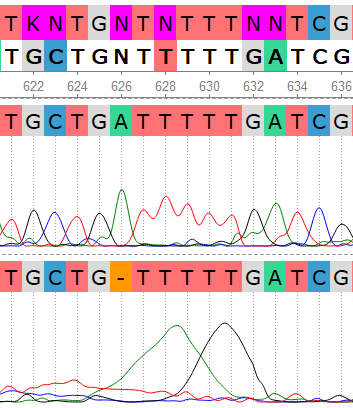

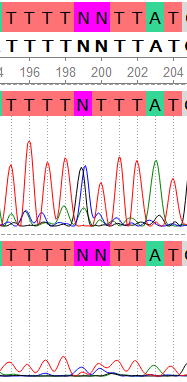
Проблемные нуклеотиды
Рассмотрим далее отдельные нуклеотиды, анализ которых вызвал наибольшие затруднения.
Здесь на позиции 199-го нуклеотида мы видим сразу три сигнала: аденин, цитозин и гуанин. Высота пика аденина небольшая, и в соответствии с тем фактом, что уровень шума на данном фрагменте достаточно заметный, можно утверждать, что этот сигнал относится к шуму и на решение влиять не будет. В обратной последовательности в данном месте тоже возникает шум, но сильнее выделяется пик цитозина. Пики цитозина и гуанина в прямой последовательности имеют примерно одинаковую форму и высоту, гуанин отсутствует в "шумовых" сигналах, что может быть основанием для признания данного явления полиморфизмом. Вероятно, произошла точечная мутация, либо исследуемый организм был гетерозиготным, и последовательности аллельных генов различаются одним нуклеотидом. Буква N в прямой последовательности была изменена на S (C/G).
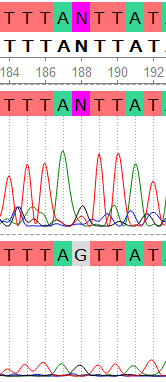
В случае 188-го нуклеотида мы наблюдаем ситуацию, когда высота первичного пика близка к минимуму и при этом присутствует выраженный шум, что вызывает сложности в прочтении. В прямой последовательности мы наблюдаем пики гуанина и тимина (гуанин более выражен), в обратной - достаточно "чистый" пик гуанина. с учетом наличия шума и соответствия уровня пика тимина уровню шума можно сделать вывод, что о полиморфизма здесь речи на идет. Прямая последовательность была отредактирована в соответствии с обратной, N заменена на G.
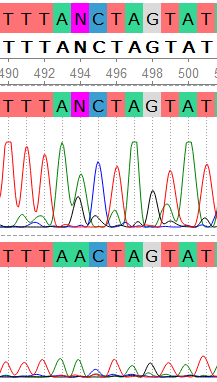
Похожая ситуация наблюдается с 494-м нуклеотидом. Мы видим два сигнала (аденин и гуанин), каждый из которых достаточно выраженный, в прямой цепи и "чистый" пик аденина в обратной. В данном фрагменте также присутствует шум, отделить который от основных пиков позволяет еще и то, что вторичные пики несколько смещены относительно первичных, их максимум не всегда лежит на центральной оси основного пика. Исходя из высота пика гуанина и общего уровня шума можно сделать вывод, что в данной позиции он должен отсутствовать. Последовательность была отредактирована в соответствии с обратной, N заменена на A.
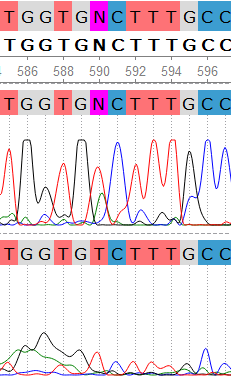
С 590-м нуклеотидом ситуация и рассуждения к ней аналогичные, пик аденина, имеющий заметно меньшую высоту и смещенный относительно центральной оси, с большой вероятностью можно посчитать шумом. Последовательность отредактирована в соответствии с обратной.
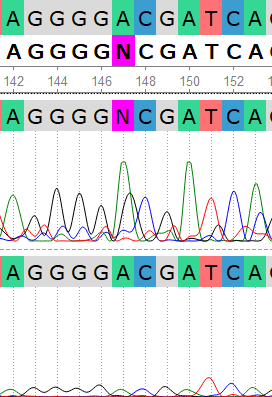
Еще один спорный момент - нуклеотид 147, на позиции которого в прямой цепи находятся пики аденина и гуанина, в обратной - пик аденина. Аналогично предыдущим рассуждениям, на участке присутствет шум, максимальные высоты "шумовых" пиков явно меньше высоты пика гуанина, который равен или даже превышает по высоте пики гуанина на соседних позициях. Несмотря на его смещение относительно центральной оси, это позволяет охарактеризовать его как полноценный вторичный пик, а не шум. Поэтому, вероятно, здесь имеет место полиморфизм. Исходя из этого решения, N в прямой последовательности была заменена на R (A/G).
Описанные и все прочие изменения отражены маленькими буквами в выравнивании полученных файлов на консенсус и отдельном fasta-файле с консенсусом, а также выделены в проекте UGENE.
Задание 2. Пример нечитаемого фрагмента хроматограммы.
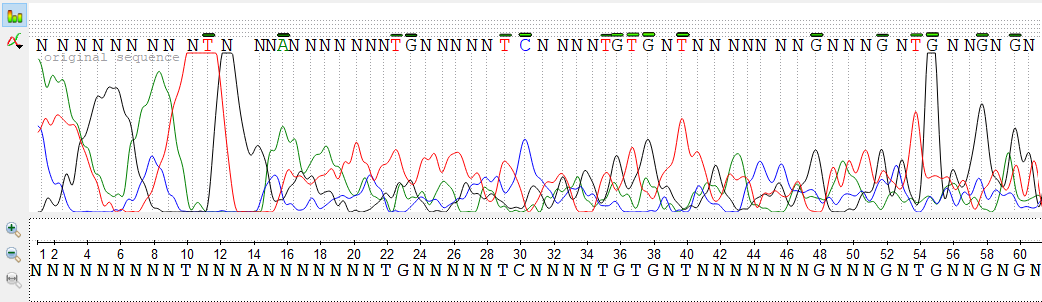
Примерами нечитаемых фрагментов могут служить начальные и конечные участки хроматограмм. На картинке выше представлен начальный участок исследуемой прямой последовательности, изображение получено также с помощью UGENE. В самом начале мы видим очень пики, накладывающиеся друг на друга и занимающие по ширине в несколько раз больше пространства, чем наблюдаемые далее, что не позволяет определить достоверно, на какой именно позиции находился нуклеотид и не обусловлена ли наблюдаемая флуоресценция ошибкой во время ПЦР. Учитывая положение фрагмента, можно предположить, что в ДНК имеются короткие неспецифические участки, на которые могут отжигаться праймеры. Далее мы видим пики более равномерные по высоте, но все еще широкие и накладывающиеся друг на друга, сигнал неотличим от шума и почти на каждой позиции можно выделить 2-3 пика, что также свидетельствует, вероятнее всего, о неспецифическом связывании праймеров и одновременной репликации разных участков ДНК.