Для работы мне были предоставлены файлы chr14.fastq и chr14.fasta.
Все действия выполнялись в специально созданной директории /nfs/srv/databases/ngs/lady_mari
Программа FastQC позволяет проводить быстрый и несложный контроль качества "сырых" данных секвенирования. Она проводит набор стандартных статистических анализов, которые можно использовать для предварительной оценки релевантности данных и выявления возможных проблем в полученной последовательности.
Использованная команда: fastqc chr14.fastq
В результате был получен архив chr14_fastqc.zip, содержащий отчет о работе программы в виде html файла - chr14_fastqc.html
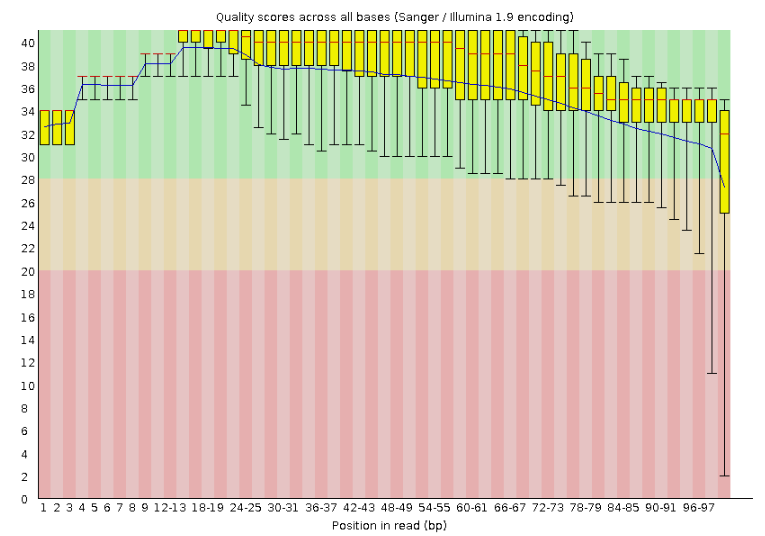
В целом качество чтений можно назвать уловлетворительным, однако для повышения качества лучше провести чистку.
Очистка чтений была проведена с помощью программы Trimmomatic. Адаптеры в исследуемой последовательности были уже удалены. Необходимо было отсечь с конца чтения нуклеотиды с неудовлетворительным качеством (< 20), для этого был указан параметр TRAILING:20. Также требовалось оставить только прочтения длины не менее 50, поэтому был указан параметр MINLEN:50.
Использованная команда:
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr14.fastq chr_trim.fastq TRAILING:20 MINLEN:50
Выходной файл:chr_trim.fastq
"Очищенная" последовательность была проанализирована с помощью FastQC.
Использованная команда:
fastqc chr_trim.fastq
Выходной файл:chr_trim_fastqc.html
до очистки
после очистки
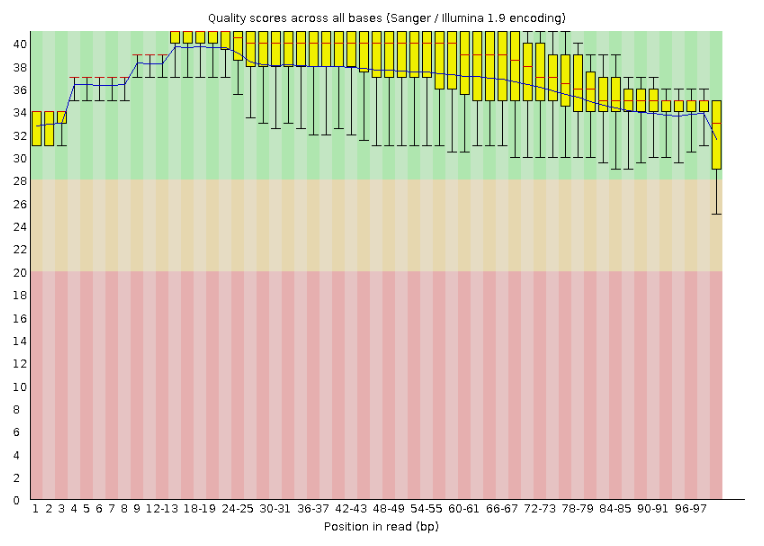
После очистки число ридов уменьшилось на 134 (с 8696 до 8562), были у ридов были удалены участки, где качество нуклеотидов ниже 20, а так же были удалены участки, длина которых меньше 50
Использованная команда:
PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
Использованная команда:
hisat2-build chr14.fasta chr14
Выход: Несколько файлов в формате ht2.
Использованная команда:
hisat2 -x chr14 -U chr_trim.fastq --no-spliced-alignment --no-softclip > 1_align.sam
Выходной файл: 1_align.sam
Использованная команда: samtools view 1_align.sam -bo 1_align.bam
Выход: 1_align.bam
Использованная команда: samtools view 1_align.sam -bo 1_align.bam
Выходной файл: 1_align.bam
Использованная команда: samtools sort 1_align.bam -T file.txt -o alignsort.bam
Выходной файл: alignsort.bam
Использованная команда: samtools idxstats alignsort.bam > reads.out
Выходной файл: reads.out
Из файла reads.out, я получила информацию, что на хромосому откартировалось 8546 чтения и 21 чтения откартированы не были.
Использованная команда: samtools mpileup -uf chr11.fasta chr11_sort.bam -o snp.bcf
Выход: snp.bcf
Использованная команда: bcftools call -cv snp.bcf -o snp.vcf
Выходной файл: snp.vcf
Было обнаружено 87 однонуклеотидных полиморфизмов и 3 инделя. Три избранных вариации охарактеризованы в таблице.
Примеры полиморфизмов Координата Тип В референсе В прочтении Качество прочтения на участке Глубина покрытия на участке 21024619 замена A G 221.999 37 21025604 вставка tg tGg 4.4191 1 21026773 замена C T 225.009 42
Далее необходимо было аннотировать полученные SNP с помощью программы ANNOVAR. Для этого сначала нужно было подготовить входной файл, чтобы программа моглас ним работать. В первую очередь из файла snp.vcf были удалены все индели, полученный файл - snp_noindel.vcf. Затем был использован скрипт convert2annovar.pl.
Использованная команда: perl /nfs/srv1/databases/annovar/convert2annovar.pl.old -format vcf4 /nfs/srv/databases/ngs/lady_mari/snp1.vcf > /nfs/srv/databases/ngs/lady_mari/snp1.avinput
Выход:snp1.avinput
Полученный файл использовался для аннотации SNP по базам данных refgene, dbsnp, 1000 genomes, GWAS и Clinvar c помощью скрипта annotate_variation.pl. В Annovar существуют 3 типа аннотаций по базам данных, основанных на: генной разметке (gene-based annotation); разметке других регионов генома (region-based annotation); фильтрации (filter-based annotation). Тип использованной аннотации указан для каждой базы данных отдельно.
Тип аннотации: gene-based annotation.
Использованная команда: /nfs/srv/databases/annovar/annotate_variation.pl.old -out rs.refgene -build hg19 snp1.avinput /nfs/srv/databases/annovar/humandb/
Полученнные файлы:
1. rs.refgene.variant_function - содержит описание всех полиморфизмов
2. rs.refgene.exonic_variant_function - содержит описание полиморфизмов внутри экзонов.
3. rs.refgene.log - содержит отчет о работе команды
База данных refseq в annovar делит snp на несколько категорий, которые отображены в первой колонке файла chr11.refgene.variant_function. Они указывают, где находится данный полиморфизм - внутри экзона, интрона, гена некодирующей РНК и т.п.
Возможные категории: exonic - полиморфизм внутри экзона (частично или полностью) splicing - полиморфизм в пределах 2 bp от границы сплайсинга (число bp можно изменить) ncRNA - полиморфизм полностью или частично входит в транскрипт, не имеющий аннотации как кодирующий UTR5 - полиморфизм полностью или частично входит в 5-нетранслируемую область UTR3 - полиморфизм полностью или частично входит в 3-нетранслируемую область intronic - полиморфизм полностью или частично внутри интрона downstream - полиморфизм в пределах 1-kb downstream от сайта окончания транскрипции (параметр может быть изменен) upstream - полиморфизм в пределах 1-kb upstream от сайта начала транскрипции (параметр может быть изменен) intergenic - полиморфизм на пересечении генов Распределение SNP по группам в моем файле: exonic intronic UTR3 Всего 3 82 1 86Если полиморфизм попадает в категории exonic/intronic/ncRNA, то во второй колонке того же файла указывается имя соответсвующего гена (если это сразу несколько генов, то они разделяются запятой). Если полиморфизм попадат в какую-либо другую категорию, то во второй колонке будут указаны два соседних гена и расстояние между ними. В моем случае в категории, для которых указывается ген, попали 85 полиморфизмов. Все они расположены на одном из трех генов - PPP2R5C, TSHR, RNASE9.
№ Координаты (*) Ген Н. замена Тип замены Качество чтений Глубина покрытия А.к. замена 1 21024619 RNASE9 A>G гомозиготная 221.999 37 S204P, S209P 2 81562998 TSHR T>C гомозиготная 222.791 18 N187N 3 102391577 PPP2R5C G>C гомозиготная 221.999 23 A546P, A476P, A515P, A531P
Тип аннотации: filter-based annotation
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.snp -build hg19 -dbtype snp138 snp1.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1.rs.snp.hg19_snp138_dropped - содержит полиморфизмы, имеющие rs в dbsnp, то есть аннотированные в этой базе данных.
2.rs.snp.hg19_snp138_filtered- содержит отфильтрованные полиморфизмы (не имеющие rs в dbsnp).
3. rs.dbsnp.log - содержит отчет о работе команды.
Аннотация по базе dbsnp показала, что 81 полиморфизмов имеют rs, а 6 не имеют.
Тип аннотации: filter-based annotation
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.snp -build hg19 -dbtype snp138 snp1.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1. rs.1000g.hg19_ALL.sites.2014_10_dropped - содержит полиморфизмы, имеющие rs в 1000 genomes, и их частоты.
2. rs.1000g.hg19_ALL.sites.2014_10_filtered -содержит полиморфизмы, не имеющие rs в 1000 genomes.
3.rs.1000.log - содержит отчет о работе команды.
Аннотация по 1000 genomes привела к схожему результату. 79 snp по-прежнему имеют rs, а 8 не имеют, ситуация с качеством и глубиной прочтения та же.
Дополнительно мы смогли узнать частоты аннотированных полиморфизмов. Они варьируют от 0,00599042
до 0,921925. Средний показатель - 0,255754918.
Тип аннотации: region-based annotation
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp1.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1.rs.gwas.hg19_gwasCatalog_dropped - содержит snp, имеющие известное клиническое значение.
Файл оказался пустым, т.е. из аннотация не дала никаких результатов.
2.rs.gwas.hg19_gwasCatalog_filtered- содержит полиморфизмы, не имеющие rs в Gwas.
3.rs.gwas.log - содержит отчет о работе команды
Тип аннотации: region-based annotation
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp1.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1.rs.gwas.hg19_gwasCatalog_dropped - содержит snp, имеющие известное клиническое значение.
Файл оказался пустым, т.е. из аннотация не дала никаких результатов.
2.rs.gwas.hg19_gwasCatalog_filtered- содержит полиморфизмы, не имеющие rs в Gwas.
3.rs.gwas.log - содержит отчет о работе команды
Тип аннотации: filter-based annotation
Использованная команда: perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.clinvar -build hg19 -dbtype clinvar_20150629 snp1.avinput /nfs/srv/databases/annovar/humandb.old/
Полученнные файлы:
1.rs.clinvar.hg19_clinvar_20150629_dropped - содержит snp, имеющие известное клиническое значение.
Файл оказался пустым, т.е. из аннотация не дала никаких результатов.
2.rs.clinvar.hg19_clinvar_20150629_filtered- содержит полиморфизмы, не имеющие rs в Clinvar.
3.rs.clinvar.log - содержит отчет о работе команды
Сводную таблицу Excel можно скачать по ссылка.
Использовались только файлы с анноированными snp (dropped).