| команда | функция |
|---|---|
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr14.1.fastq chr14.1_trim.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| hisat2 -x chr14 -U chr_trim.fastq --no-softclip > 14.1_align.sam | Создание выравнивания референсной последовательности и прочтений (был убран параметр "--no-spliced-alignment", т.к. выравнивают транскипты с референсной последовательностью) |
| samtools view 14.1_align.sam -bo 14.1_align.bam | Перевод выравнивания в бинарный формат |
| samtools sort 14.1_align.bam -T file.txt -o 14.1_alignsort.bam | Сортировка бинарного выравнивания |
hisat2 --no-softclip -x chr14 -U chr14.1_trim.fastq -S 14.1_align.sam &> result.log | Выравнивает риды с референсной последовательностью, сохраняя информацию о работе программы в файл result.log.
Из файла result.log можно узнать следующие данные о картировании. 18134 было непарных ридов, 47 не были картированы, 18087 были картированы ровно 1 раз, 0 картировано больше одного раза
htseq-count -f 14.1_bam align.bam -m -i gene_id -s /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf | Посчитывает, сколько раз на ген попали последовательности из ридов с различными параметрами.
вывод программы:
__no_feature 5266
__ambiguous 0
__too_low_aQual 0
__not_aligned 20
__alignment_not_unique 8
grep -v '0$' count.out > ww | Найти все гены, на которые лег хотя бы 1 рид.
вывод программы:
ENSG00000078304.15 2057 ENSG00000165409.11 372 ENSG00000188655.6 325 ENSG00000258915.1 336 ENSG00000259161.2 27 ENSG00000259167.2 87 ENSG00000271656.1 18 ENSG00000271705.1 52
Опции:
-f или --format: формат файла (.bam или .sam) с выравниванием (по умолчанию sam)/
-i или --idattr: какой использовать атрибут GFF в качестве feature ID (по умолчанию gene_id).
-s или --stranded: yes - считать только по прямой цепи, no - считать без учета цепи, reverse - считать только по обратной полследовательности.
-m или --mode: определяет, как считать различное наложение рида на гены в том случае, когда это неоднозначно. Есть 3 режима: union, intersection-strict и intersection-nonempty. Риду присваивается свойство в зависимости от режима работы. Свойство no_feature означает, что рид не попал ни на один ген, свойство ambiguous - рид попадает на несколько генов и не может однозначно быть соотнесён с конкретным геном.
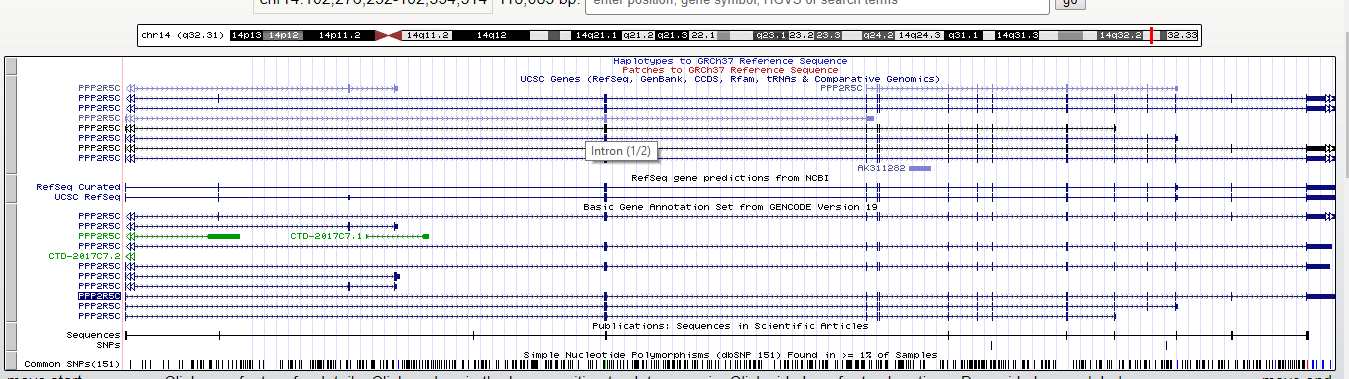
Обзор самого покрытого гена ENSG00000078304.15>
Это ген. кодирующий белок человеческой фосфотазы 2.
Строение гена из базы данных UCSCБ