Сравнение выравниваний одних и тех же последовательностей разными программами
Изначально для анализа были взяты цитохромы С разных организмов, однако их высокая эволюционная консервативность и небольшая длина не позволили выявить значимых различий в работе разных программ для выравниваний. Следующим было выбрано семейство глицеральдегид-3-фосфат дегидрогеназ (мнемоника G3P). Глицеральдегид-3-фосфат дегидрогеназа – один из ключевых ферментов гликолиза, однако наличие нескольких структурных доменов и функциональных петель делает ее последовательность не слишком консервативной. Такая мозаичность структуры должна помочь эффективно выявить различия в эвристических методах, используемых программами Muscle, Mafft и Clustal Omega. При этом белки хорошо изучены и представлены почти во всех группах живых организмов.
Поиск производился по запросу «(id:G3P_*) AND (reviewed:true)» в UniProt. Для анализа были выбраны записи с ID G3P_HUMAN, G3P_MOUSE, G3P_MESAU, G3P_SHEEP, G3P_BOLED, G3PC_TRYBB, G3P_KLEPN, G3P_SACS2, соответствующие G3P-дегидрогеназам человека, мыши, хомяка, овцы, белого гриба, трипаносомы, клебсиеллы и термофильной археи.
Выравнивания, проведенные программами Muscle, Mafft и ClustalO через Jalview сравнивались с помощью программы macho, написанной прошлым поколением первокурсников. Результаты работы программы приведены в таблицах 1 и 2.
Ссылка на скрипт (сверху есть указания на авторов)
Гиперссылка на файл с проектом Jalview
Выравнивание Muscle в формате fasta
Выравнивание Mafft в формате fasta
Выравнивание ClustalO в формате fasta
| № Совпадающего участка | Координаты (Muscle) = (Mafft) | Длина участка |
|---|---|---|
| 1 | (1, 46) = (1, 46) | 46 |
| 2 | (52, 53) = (52, 53) | 2 |
| 3 | (97, 97) = (111, 111) | 1 |
| 4 | (110, 110) = (124, 124) | 1 |
| 5 | (115, 122) = (129, 136) | 8 |
| 6 | (127, 127) = (141, 141) | 1 |
| 7 | (130, 131) = (144, 145) | 2 |
| 8 | (134, 245) = (148, 259) | 112 |
| 9 | (287, 299) = (324, 336) | 13 |
| 10 | (322, 322) = (359, 359) | 1 |
| 11 | (346, 352) = (383, 389) | 7 |
| № Совпадающего участка | Координаты (Muscle) = (ClustalO) | Длина участка |
|---|---|---|
| 1 | (1, 34) = (1, 34) | 34 |
| 2 | (40, 49) = (40, 49) | 10 |
| 3 | (52, 53) = (52, 53) | 2 |
| 4 | (90, 96) = (95, 101) | 7 |
| 5 | (98, 100) = (103, 105) | 3 |
| 6 | (102, 108) = (107, 113) | 7 |
| 7 | (111, 125) = (116, 130) | 15 |
| 8 | (127, 128) = (132, 133) | 2 |
| 9 | (131, 132) = (136, 137) | 2 |
| 10 | (134, 145) = (139, 150) | 12 |
| 11 | (148, 214) = (153, 219) | 67 |
| 12 | (217, 218) = (222, 223) | 2 |
| 13 | (221, 273) = (226, 278) | 53 |
Выдача Muscle была выбрана в качестве референсной. Выравнивание через Mafft вышло самым растянутым (411 позиций против 372 у Muscle), из-за чего процент совпадающих колонок у него оказался самым низким – 47.20%, а суммарная длина совпавших участков составила 194 аминокислоты. На старте алгоритм MAFFT дольше удерживал совпадение, но затем показал резкие скачки и крупноблочные сдвиги. Алгоритм ClustalO оказался больше похож на Muscle: итоговая длина его выравнивания составила 386 позиций и процент совпадающих колонок с Muscle заметно выше – 55.96%. При этом суммарная длина совпавших участков у ClustalO составила 216 позиций. В отличие от Mafft, ClustalO распределяет гэпы более равномерно, блоки смещаются без резких координатных разрывов. Стоит отметить, что все три программы одинаково выровняли начало (позиции 1–34) и крупный блок в центре (позиции 148–214 в Muscle), что указывает на высокую эволюционную стабильность этих участков и достоверность выравниваний в их пределах.
Сравнение выравнивания по совмещению структур с выравниванием Muscle
Для дальнейшего анализа были выбраны 3 белка c TEA-доменом из семейства Pfam PF01285: 2hzd, 5gzb и 5nnx. Сначала белки были выравнены с помощью Muscle, затем через PDBeFold. Выравнивания сравнивались ручным методом в Jalview. Результаты сравнения приведены в таблице 3.
Гиперссылка на файл с проектом Jalview
Выравнивание Muscle в формате fasta
Выравнивание PDBeFold в формате fasta
| Номер участка | Тип участка | Muscle и PDBeFold (координаты) | Muscle и PDBeFold (длина) |
|---|---|---|---|
| 1 | совпадение | (1,25)=(1,25) | 25 |
| 2 | несовпадение | (26,36)=(26,33) | 11/8 |
| 3 | совпадение | (37,39)=(34,36) | 3 |
| 4 | несовпадение | (40,42)=(37,38) | 3/2 |
| 5 | совпадение | (43,59)=(39,55) | 17 |
| 6 | несовпадение | (60,65)=(56,60) | 6/5 |
| 7 | совпадение | (66,80)=(61,75) | 15 |
| 8 | несовпадение | (81,85)=(76,79) | 5/4 |
| 9 | совпадение | (86,104)=(80,98) | 19 |
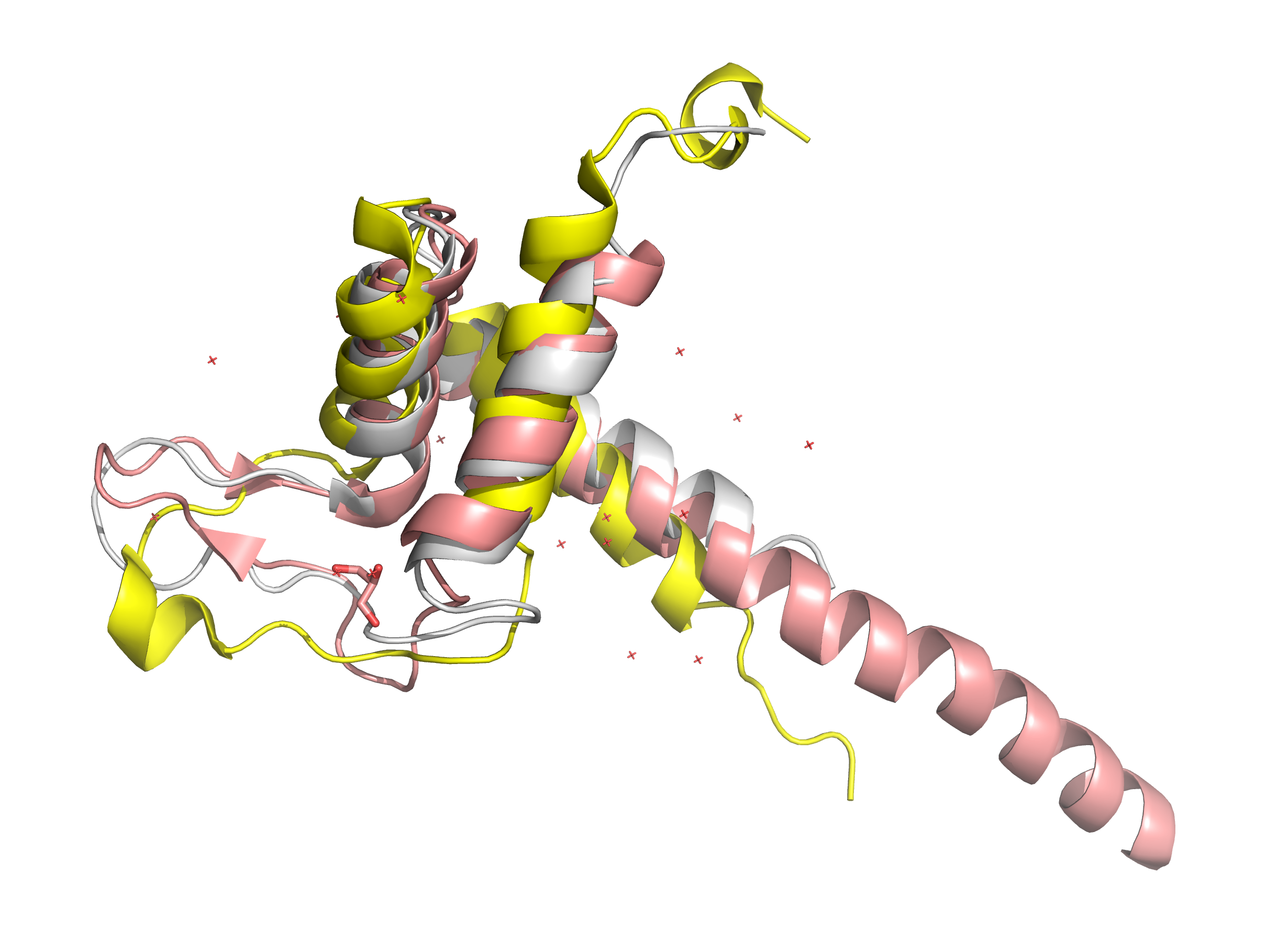
Сравнение выравниваний MUSCLE и PDBeFold показало 5 совпадающих и 4 несовпадающих участка. Совпадения приходятся на структурно консервативные области (α-спирали и β-листы), несовпадения – на петли и вариабельные участки. Это связано с разными принципами работы методов: MUSCLE выравнивает по первичной последовательности, PDBeFold – по пространственной структуре.
Краткое описание программы Muscle
MUSCLE (MUltiple Sequence Comparison by Log-Expectation) – одна из популярных программ для множественного выравнивания белков и нуклеиновых кислот. Создатель алгоритма – американский биоинформатик Роберт С. Эдгар [1].
Основные возможности[1]:
Как и другие программы для выравнивания, MUSCLE предназначена для сопоставления нескольких аминокислотных или нуклеотидных последовательностей для поиска эволюционно или структурно значимых участков. Программа способна выравнивать тысячи последовательностей за минуты на обычном компьютере, превосходя старые алгоритмы по качеству итогового выравнивания. Также MUSCLE дает пользователю возможность выбрать подходящий режим (настроить точность выравнивания, скорость обработки данных или взять вариант по умолчанию).
Алгоритм Muscle [1]:
Используется метод прогрессивного выравнивания с последующим уточнением. Проходит в три основных этапа.
- Быстрая оценка расстояний. На основе подсчета совпадающих коротких фрагментов (k-mer) строится начальное дерево гипотетического родства. Преимущество в том, что не нужно проводить множество попарных выравниваний.
- Прогрессивное выравнивание. Построенное дерево используется для объединения последовательностей в профили, которые выравниваются между собой с использованием функции оценки log-expectation.
- Стадия уточнения. Алгоритм делит на части полученное выравнивание, перевыравнивает их и сохраняет результат, если итоговый балл (SP-score) улучшился. Это позволяет исправлять ошибки, допущенные на ранних этапах.
В 2022 году вышла MUSCLE v5, которая стала использовать скрытые марковские модели (HMM), что сделало алгоритм точнее и позволило лучше оценивать достоверность [2].
Список литературы
[1] Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32(5): 1792–1797. https://academic.oup.com/nar/article/32/5/1792/2380623
[2] Edgar RC (2022) Muscle5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nature Communications 13: 6968. https://www.nature.com/articles/s41467-022-34630-w