Задания практикума 15 (cборка de novo)
Подготовка чтений программой trimmomatic.
Удалим адаптеры. Программа trimmomatic:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240379.fastq SRR4240379_trimm.fastq ILLUMINACLIP:adapters.fasta:2:7:7
После триммирования было убрано 1.76% процента от входной последовательности. Input Reads: 7400155 Surviving: 7269852 (98.24%) Dropped: 130303 (1.76%)
Удаление с правых концов чтений нуклеотиды с качеством ниже 20, удаление чтений, длина которых меньше 32 нуклеотидов..
java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240379_trimm.fastq SRR4240379_outtrimm.fastq TRAILING:20 MINLEN:32
После этого было удалено 4.07% от входной последовательности.
Input Reads: 7269852 Surviving: 6974267 (95.93%) Dropped: 295585 (4.07%) TrimmomaticSE: Completed successfully
Размеры файлов: изначально: 782194222, после первого триммирования: 768146676, после второго: 735535926.
Используем программу velveth
velveth velvet/ 31 -short -fastq SRR4240379_outtrimm.fastq
Здесь velvet/ — директория для сборки, 31 — размер k-мера для построения графа, -short — указание на короткие непарные прочтения.
velvetg . -cov_cutoff auto
Final graph has 75 nodes and n50 of 27786, max 49912, total 655105, using 0/6974267 reads
Получила таблицу со всеми прочтениями. Проанализировала ее и нашла самые длинные прочтения
ТаблицаДля удобства каждое из трех прочтений я вывела в отдельный файл: прочтение 5 , прочтение 6 , прочтение 9.
Таблица:cписок и характеристики самых длинных прочтений.
| ID | Длина | Покрытие |
| 6 | 49912 | 35.6 |
| 9 | 49262 | 34.8 |
| 5 | 33085 | 36.3 |
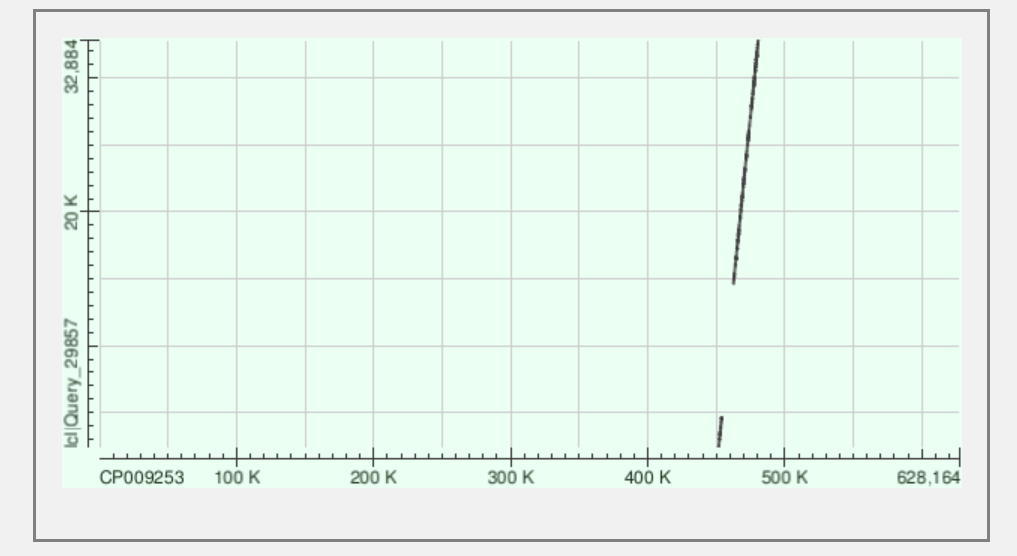
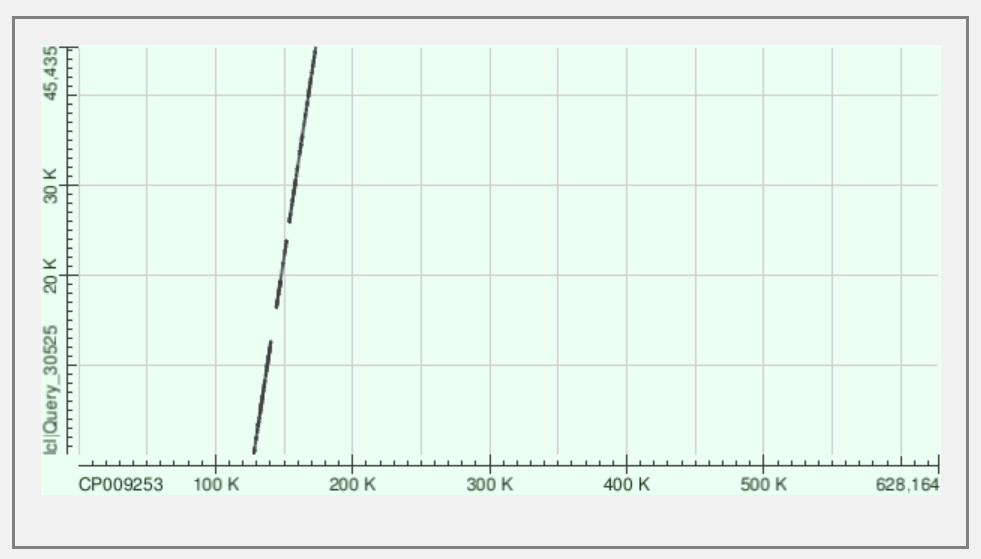
Blast с CP009253:Buchnera aphidicola
| 6 | 9 | 5 | |
| Координаты хромосомы | 127825 - 173180 | 500370 - 529211 | 467412-480660 |
| Score | 5421 | 10336 | |
| Идентичность | 75% | 76% | 77.03% |
| Gaps | 4% | 4% | 2% |
| E-value | 0.0 | 0.0 | 0.0 |
Я указала координаты области в геноме бактерии, на которую выравниваются прочтения. Не указывала подробно координаты каждой части выровненного прочтения. Эту информацию при желании можно достать из blast. Ниже привожу на картинки, на которых видно, что прочтения выровнялись не ровно, а с инделями. Где-то маленькими, где то существенно крупными. Это ожидаемо, так как мы осуществляем сборку нового генома, выравниваем на родственный организм.