Миниобзор генома бактерии Streptomyces rutgersensis
Черных Алексей Вячеславович
Факультет биоинженерии и биоинформатики Московского государственного университета имени М.В. Ломоносова
Аннотация
Биотехнология играет ключевую роль в современном мире, обеспечивая решения для многих глобальных проблем, таких как продовольственная безопасность, получение лекарств и вакцин, разработка новых экологически чистых технологий. В данной работе нами были описаны некоторые характеристики генома и протеома бактерии Streptomyces rutgersensis, а также проведён анализ полученных данных с проверкой гипотез. Основной темой работы стало выявление общих паттернов и индивидуальных особенностей, которые могут использоваться в биотехнологии для решения широкого круга задач по модификации данной бактерии.
Введение
Streptomyces rutgersensis относится к грамположительным бактериям класса актинобактерий, порядка Streptomycetales семейства Streptomycetaceae [1].
Согласно последним исследованиям о реклассификации представителей рода Streptomyces: Streptomyces rutgersensis и Streptomyces gougerotii классифицируются как более поздние гетеротипические синонимы S. diastaticus [1].
Актинобактерии населяют почвы, а также пресные и морские воды. Они играют важную роль в разложении органических веществ, таких, как целлюлоза и хитин, и таким образом принимают участие в круговороте органических веществ и в углеродном цикле [2].
Для Streptomyces rutgersensis характерна ферментативная активность по разложению лигноцеллюлозы. Одним из выделяемых ими ферментов является ксиланаза. Другие исследования показали значительную эффективность использования данного вида при отбеливании бумажной массы [2].
Использование микроорганизмов в целлюлозно-бумажной промышленности доказало, что технология биоотбеливания является экологически чистой альтернативой традиционному подходу.
Материалы и методы
Во время исследования генома и протеома бактерии Streptomyces rutgersensis нами были использованы нуклеотидные последовательности CDS генов бактерии и таблица локальных особенностей, которые были загружены из банка NCBI (Сопроводительные материалы [1]).
Для анализа длин белков, закодированных в геноме бактерии, была построена гистограмма в электронных таблицах Google Sheets (Сопроводительные материалы [2]). Данные были взяты из таблицы локальных особенностей, загруженной и импортированной ранее в лист cds, где у неё были удалены некоторые неиспользуемые столбцы (Сопроводительные материалы [3]). На листе prot_len_hist были найдены абсолютные значения длин белков, а также подсчитано среднее для них. После этого было получено количество белков, входящих в определенные до этого карманы. На основе полученной таблицы была построена гистограмма.
Оценка количества генов белков и генов разных типов РНК для каждого репликона осуществлялась перебором строк таблицы локальных особенностей и распределением их на 2 группы: NZ_CP045704.1 (хромосомные) и NZ_CP045705.1 (плазмидные). Для выполнения данной задачи был написан скрипт для командной оболочки Bash (Сопроводительные материалы [4]). На вход ему подаётся таблица локальных особенностей, в результате её перебора на выход выдаётся таблица. В первом столбце указано количество генов, во втором их тип, а в третьем имя репликона, на котором они содержатся. Для удобства восприятия, в текст обзора была вставлена переработанный вариант полученной таблицы.
Для оценки GC состава CDS бактерии была построена гистограмма зависимости количества CDS от содержания GC % в них. Для начала с помощью написанного скрипта для интерпретатора Python для каждого CDS из fasa-файла было подсчитано количество G и C нуклеотидов, а также общая длина (Сопроводительные материалы [4]). Данные выдаются в два массива соответсвенно. После этого они были занесены в столбцы листа электронной таблицы, где подсчитано содержание GC в % для каждого CDS, а также проведено их разбиение по карманам (Сопроводительные материалы [5]). На основе данных получившейся таблицы была построена гистограмма.
Встречаемость старт-кодонов CDS для бактерии вычислялась путём перебора нуклеотидных последовательностей. Для этого был написан скрипт для командной оболочки Bash (Сопроводительные материалы [6]). На вход ему подаётся fasta-файл , в результате его перебора на выход выдаётся таблица. В первом столбце содержится количество встреченных старт-кодонов, а во втором их тип.
Проверка зависимости между длинной CDS и типом её старт-кодона была проведена с помощью дисперсионного анализа. Для этого был написан скрипт для интерпретатора Python с помощью библиотеки scipy.stats (Сопроводительные материалы [7]).
Результаты
Длины белков, закодированных в геноме бактерии Streptomyces rutgersensis
На рисунке 1 приведено распределение длин белков. Наибольшее количество белков имеет длину 200–300 аминокислотных остатков. Далее с увеличением длины количество белков убывает. Минимальная длина белка — 18 ам. ост. , а максимальная — 10649 ам. ост. Средняя длина белка — 337 аминокислот.
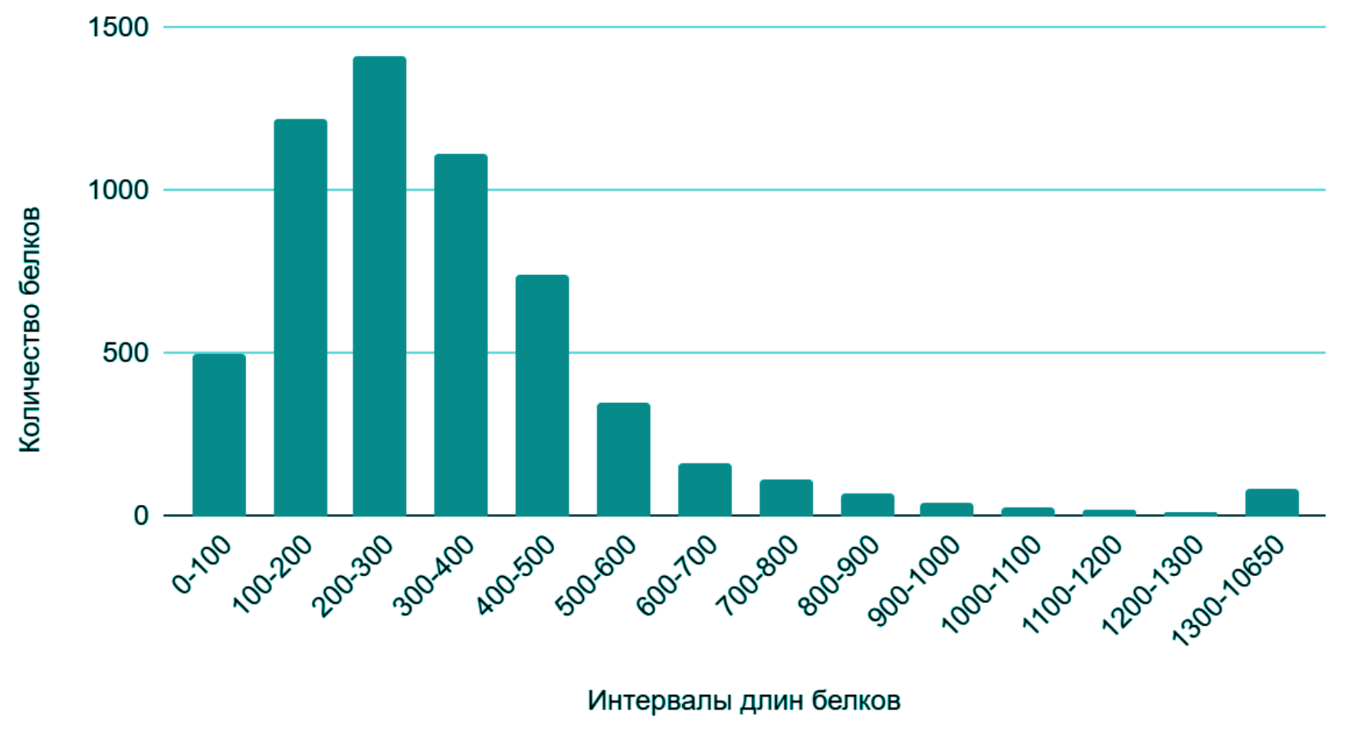
Рисунок 1. Гистограмма длин белков
Длина синтезируемых белков может быть обусловлена множеством факторов, среди них нами были выделены следующие:
1. Изменения условий обитания
Если бактерия живёт в нестабильных условиях и ей требуется быстрое приспособление к ним, то в геноме могут преобладать гены, кодирующие короткие белки. Это обусловлено тем, что их можно быстро транскрибировать и транслировать.
2. Ограничение ресурсов
Средняя длина белков может отражать ограничения, обусловленные доступностью необходимых ресурсов (например, аминокислот). В случае их нехватки длина большинства белков будет стремиться к уменьшению. Из гистограммы видно, что белки распределены не равномерно на интервале от 0 до 10650 аминокислот. При этом большинство из них расположены в его меньшей половине, что говорит о преобладании коротких белков над длинными. Это может свидетельствовать о быстрой приспособляемости данной бактерии к условиям среды обитания, а также её нетребовательности к наличию большого количества ресурсов. Оба этих качества являются преимущественными при выборе объекта для использования в биотехнологии.