Учебный сайт Фоменко Елены
| Главная | Семестры | Проекты | Заметки |
Цель занятия - ознакомится с возможностями докинга низкомолекулярного лиганда в структуру белка.
Будем работать с белком лизоцимом, структуру которого строили на основе гомологичного моделирования на прошлом практикуме.
Программе Autodock Vina для докинга необходимы специально форматированные файлы pdb c зарядами и указанием торсионных углов.
Для начала попробуем провести докинг одного из мономеров сахара (NAG) из прошлого занятия.
1. В банке pdb находим SMILES нотацию для NAG. Сохраняем эту нотацию в файл nag.smi.
2. C помощью obgen строим 3D структуру этого сахара в pdb формате.
Использовались команды:
obgen nag.smi > nag.mol
export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Полученную структуру через PyMol сохраняем как .pdb.
Скриптом prepare_ligand4.py из пакета Autodock tools создаем pdbqt файл лиганда. Указываем для этого путь к скрипту:
export PATH=${PATH}:/home/preps/golovin/progs/bin
Так же, скриптом prepare_receptor4.py из пакета Autodock tools создаем pdbqt файл белка.
Теперь создаем файл с параметрами докинга vina.cfg. Для докинга необходимо указать область структуры белка, в которой будет происходить поиск места для связывания. Задаем ее как куб с неким центром. Координаты центра определяем из модели комплекса, которую мы построили на прошлом занятии. Определили центр масс с помощью PyMol, командой pseudoatom, координаты вывели командой iterate_state 1, pseudo01, print x,y,z.
Теперь проводим первый докинг:
vina --config vina.cfg --receptor protein.pdbqt --ligand nag.pdbqt --out nag_protein.pdbqt --log nag_prot.log
Получаем nag_prot.log, ниже представлены энергии трех лучших расположений и геометрическая разница между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.2 0.000 0.000
2 -5.1 2.375 4.295
3 -5.1 2.021 3.280
В PyMol загружаем файлы nag_protein.pdbqt и protein.pdbqt. Отображаем все состояния на одной картинке с помощью split_states (Рис.1).
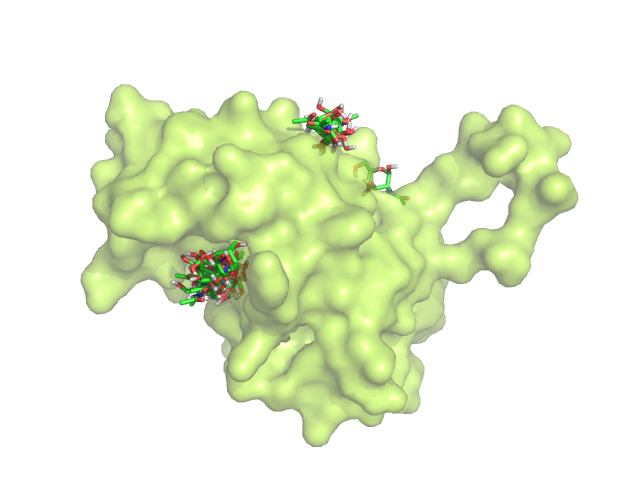
Рис.1 Изображение всех состояний после первого докинга.
Оказалось, что лиганд связывается чаще всего именно в предусмотренном для него месте.
Теперь проведём докинг, рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты, которые мы использовали в прошлом задании для позиционирования лиганда.
python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r protein.pdbqt -s ASN87_GLN145_LYS147
и проведём докинг:
vina --config vina.cfg --receptor protein_rigid.pdbqt --flex protein_flex.pdbqt
--ligand nag.pdbqt --out nag_prot_flex.pdbqt --log nag_prot_flex.log
Получаем файл nag_prot_flex.log, ниже представлены энергии трех лучших расположений и геометрическая разница между ними.
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.8 0.000 0.000
2 -5.3 2.221 4.219
3 -5.2 1.543 3.742
Энергии получились немного лучше, чем в первом случае, а времени затрачено немного больше.
Загружаем в PyMol nag_prot_flex.pdbqt и protein_rigid.pdbqt,
отображаем все состояния на одной картинке (Рис.2).
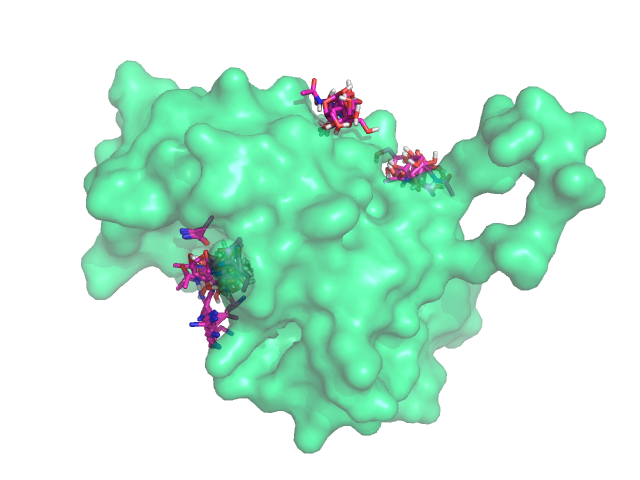
Рис.2 Изображение всех состояний после второго докинга.
Заметно, что в "кармане" лиганды стали располагаться менее централизованно (видимо, как следствие подвижности некоторых остатков в том районе), и больше лигандов связалось с другой стороны белка.
NAG содержит в себе СH3C(=O)NH группу. Создаем 4 лиганда, где метильный радикал этой группы будет заменён на OH, NH2, H или Ph:
nagoh.smi
nagnh2.smi
nagh.smi
nagph.smi
Для каждого из этих лигандов проводим обыкновенный докинг.
Таблица из трёх лучших расположений для каждого лиганда:
nagoh_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.1 0.000 0.000
2 -4.0 4.286 6.153
3 -3.8 2.962 4.195
nagnh2_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.4 0.000 0.000
2 -4.3 1.889 3.883
3 -4.2 4.571 6.855
nagh_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.3 0.000 0.000
2 -4.1 3.824 5.539
3 -4.1 1.000 2.914
nagph_prot.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.7 0.000 0.000
2 -5.6 2.318 2.996
3 -5.4 2.393 3.986
Можно заметить, что энергии лучших расположений ухудшились во всех случаях (в меньшей степени для Ph-замещенного лиганда, в большей степени для OH-замещенного).
Проведем теперь докинг с подвижными радикалами для новых 3 лигандов, кроме Н. Энергии лучших расположений:
nagoh_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -4.5 0.000 0.000
2 -4.4 1.442 3.515
3 -4.3 1.475 2.924
nagnh2_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -5.1 0.000 0.000
2 -5.1 11.632 13.795
3 -5.1 12.445 14.445
nagph_prot_flex.log
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -6.3 0.000 0.000
2 -6.3 5.539 9.082
3 -6.1 2.185 5.905
Энергии получились лучше, чем в предыдущем случае, а для Ph-замещенного лиганда даже лучше, чем при обычном докинге незамещенного лиганда. Видимо, при определенных конформациях белка этот лиганд обладает немного повышенным сродством.