Учебный сайт Фоменко Елены
| Главная | Семестры | Проекты | Заметки |
1. Поиск ДНК-белковых контактов в заданной структуре.
Упр.1
Предстоит вспомнить, как с помощью команды define RasMol задавать множества атомов.
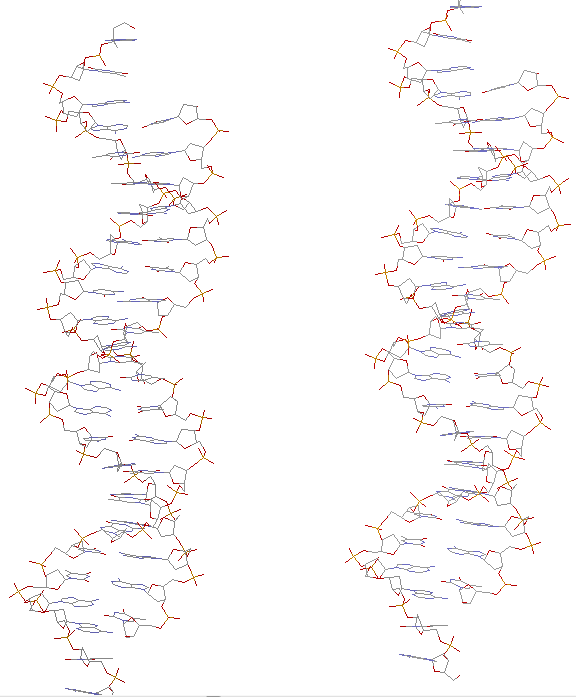
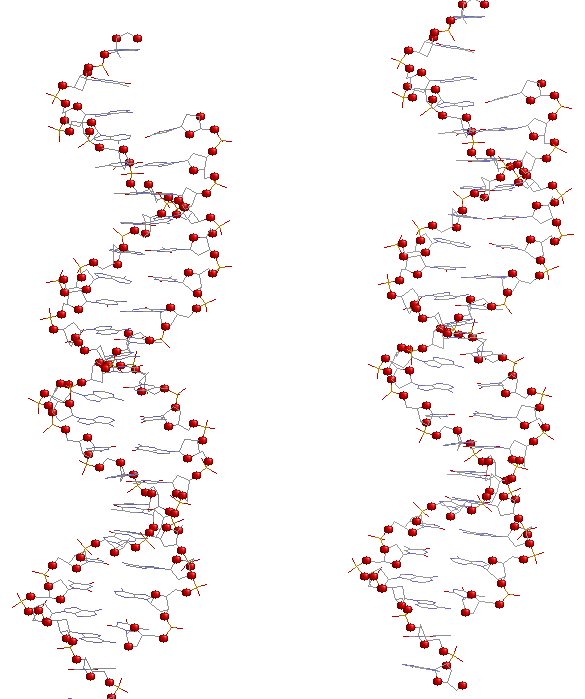
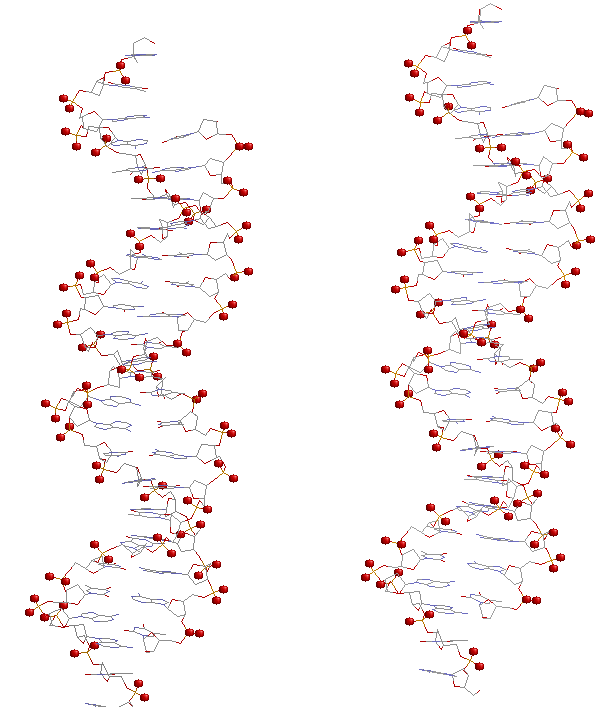
1. Множество атомов кислорода 2'-дезоксирибозы (set1).
2. Множество атомов кислорода в остатке фосфорной кислоты (set2).
3. Множество атомов азота в азотистых основаниях (set3).
4. Скрипт-файл с определениями этих множеств.
5. Скрипт-файл, вызов которого в RasMol дает последовательное изображение всей структуры, только ДНК в проволочной модели, той же модели,
но с выделенным шариками множеством set1, затем set2 и set3.
Упр.2.
Будем описывать ДНК-белковые контакты в заданной структуре и сравнивать количество контактов разной природы.
Считаем полярными атомы кислорода и азота, а неполярными - атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5A. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии < 4.5A.
Таблица "Контакты разного типа в комплексе 1_KSX.pdb".
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 4 | 89 | 93 |
| остатками фосфорной кислоты | 74 | 96 | 170 |
| остатками азотистых оснований со стороны большой бороздки | 0 | 59 | 59 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 2 | 2 |
Белок больше всего контактирует с остатками фосфорной кислоты ДНК. Неполярные контакты преобладают. Мало контактов у белка с остатками фзотистых оснований со стороны малой бороздки.
Был использован скрипт, определяющий множества атомов.
Упр.3
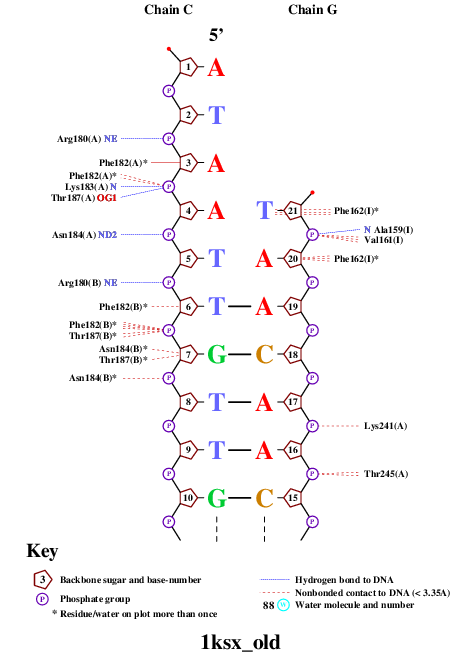
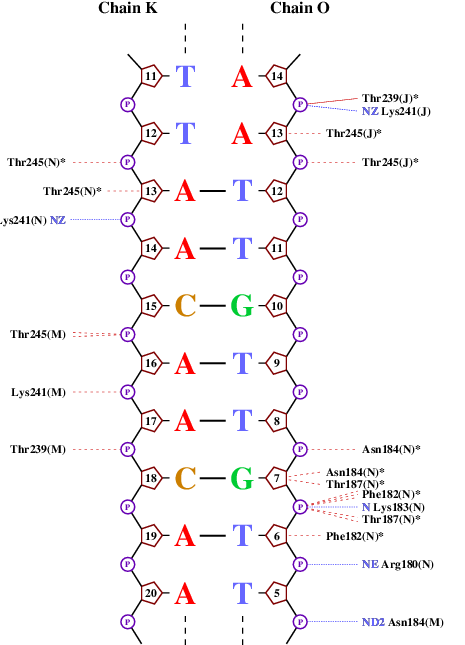
Получим популярную схему ДНК-белковых контактов с помощью программы nucplot.
Программа nucplot, предназначенная для визуализации контактов между ДНК и белком, запускается на сервере kodomo. Программа работает только со старым форматом PDB.
Получаем изображения (для одной ДНК):
Упр.4
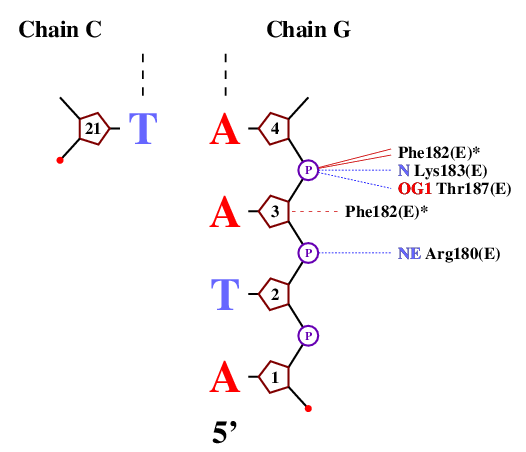
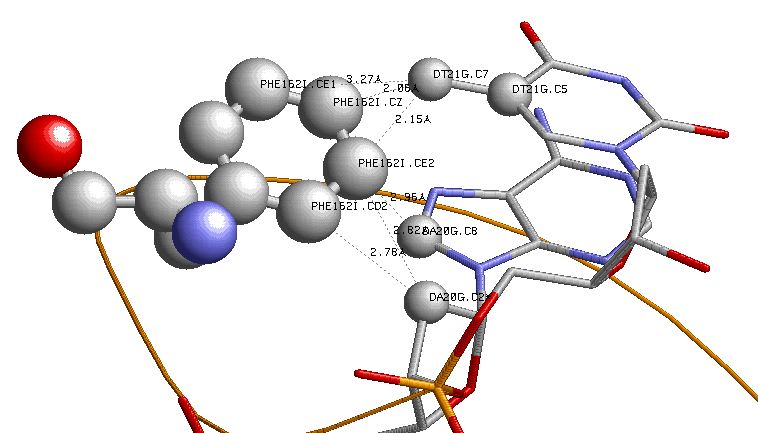
a. Аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК - Phe162(I), образующий 6 контактов.
b. По-моему мнению, наиболее важный АО для распознавания последовательности ДНК - Phe162(I), связанный непосредственно с азотистым основанием Т.
В однажды полученном файле 1ksx_old.bond находим:
**** Non Bonded Contacts **********************
protein DNA Distance
PHE I 162 CD2 A G 20 C2* 2.78
PHE I 162 CE2 A G 20 C2* 2.83
PHE I 162 CE2 A G 20 C8 2.96
PHE I 162 CE2 T G 21 C5 3.17
PHE I 162 CE1 T G 21 C5M 3.28
PHE I 162 CE2 T G 21 C5M 2.16
PHE I 162 CZ T G 21 C5M 2.07
Смотрим эти контакты с помощью RasMol (подписаны атомы, участвующие во взаимодействиях):
2. Предсказание вторичной структуры заданной тРНК
Упр.1
Будем предсказывать вторичную структуру тРНК путем поиска инвертированных повторов.
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях.
Находим возможные комплементарные участки в последовательности исследуемой тРНК.
При стандартных параметрах получила пустые файлы. Уменьшив "Minimum score threshold" до 10, получила на выходе один только стебель, акцепторный,
и он на одну пару нуклеотидов длиннее, чем полученный find-pair. Вообще, каким-то образом, нумерация в out-файле, полученном после find-pair,
совсем не соответствует pdb-файлу и отдельно последовательности; что-то с ним не то.
Меняем параметры дальше:
Gap penalty [12]: 8 Minimum score threshold [50]: 10 Match score [3]: 10 Mismatch score [-4]: -2
Нашелся акцепторный стебель и неточно - антикодоновый.
Gap penalty [12]: 1 Minimum score threshold [50]: 5 Match score [3]: 5 Mismatch score [-4]: -1
Нашлись антикодоновый и акцепторный стебли, похоже на реальность.
После перебора множества других вариантов параметров ничего более удачного уже не получилось, больше никаких стеблей не нашлось.
Упр.2
Будем предсказывать вторичную структуру тРНК по алгоритму Зукера.
Использовался web-вариант.
При параметрах по умолчанию структура получлась слишком далекой от реальности.
Меняем параметр P. Он указывает, на сколько процентов выдаваемое предсказание структуры может отличаться по своей вычисленной энергии от оптимального.
Чем больше значение этого параметра, тем больше вариантов предсказания будет выдано.
При изменении этого параметра с 5 на 10 мало что изменилось, хоть и получилось 2 варианта структуры на выходе.
Но при P=20 последняя из 4 полученных структур оказалась вполне правдоподобной:
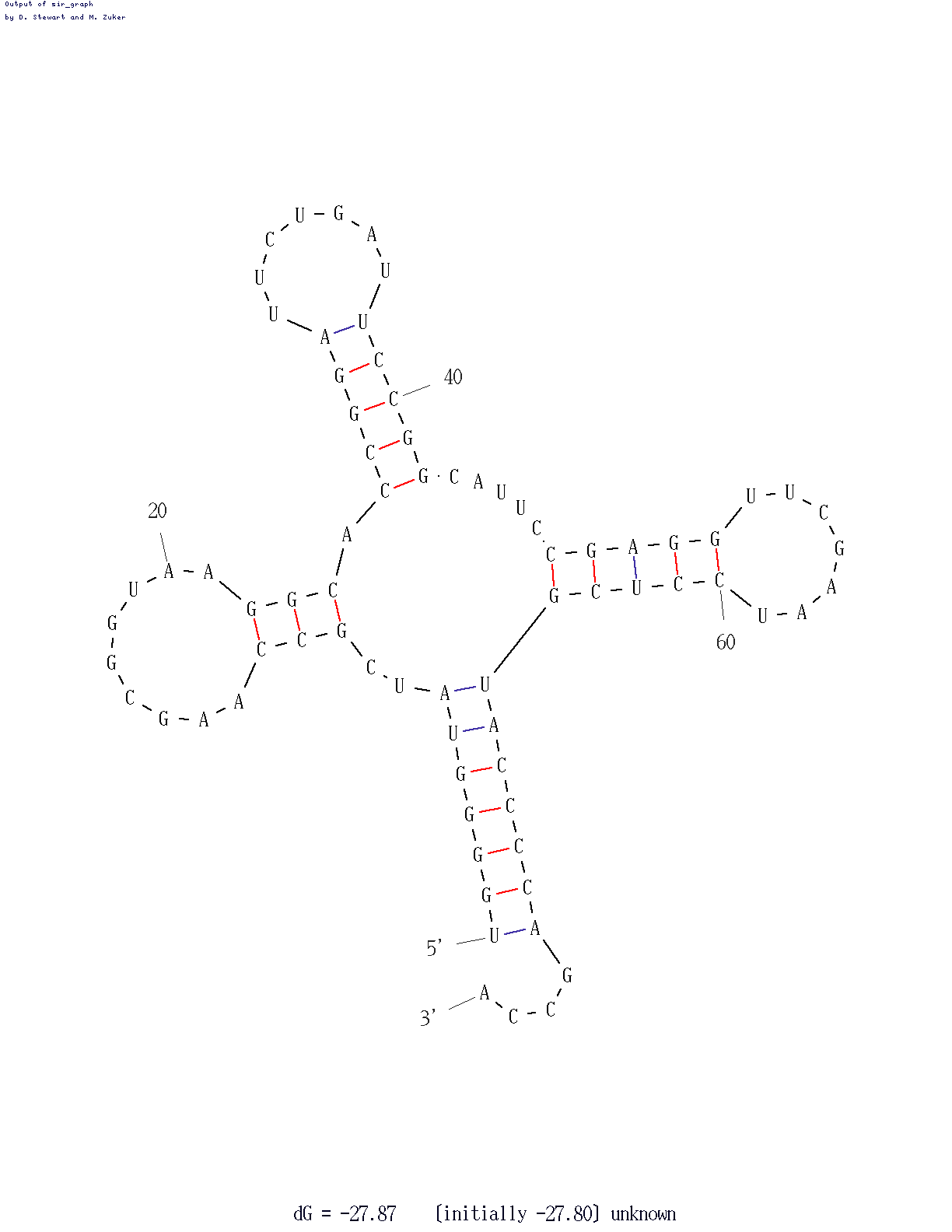
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель |
5'-2-7-3' 5'-66-71-3' Всего 6 пар (все канонич.) |
все 6 + 1 |
все 7 канонич. |
| D-стебель | 5'-10-12-3' 5'-23-25-3' Всего 3 пары (все канонич.) |
Не найдено | все 3 |
| T-стебель | 5'-49-53-3' 5'-61-65-3' Всего 5 (все канонич.) |
Не найдено | Все 5 |
| Антикодоновый стебель | 5'-37-44-3' 5'-26-33-3' 9 пар (6 канонических) |
все 6 + 1 | 5 канонич. |
| Общее число канонических пар нуклеотидов | 20 | 14... | 20 |