1School of Bioengineering and Bioinformatics, Lomonosov Moscow State University
Abstract
Klebsiella variicola is a species of bacteria which was originally identified as a benign
endosymbiont in plants, but has since been associated with disease in humans, animals. Thus,
research concerning the protein profile of this bacteria is essential for understanding its
pathogenicity. This investigation characterized key genomic and proteomic features,
determining: protein length distribution; the number of protein-coding and various RNA genes
encoded on every replicon; genomic GC content; the distribution of intergenic gaps lengths;
and the locations of the replication origin (oriC) and terminus (terC). Also, the comparison of
K. variicola’s genome with different Klebsiella species was carried out.
1 Introduction
Klebsiella variicola was described as a species of Klebsielladistinct from its
closely related species Klebsiella pneumoniae in 2004. Like other Klebsiella
species, K.variicola is gram-negative, rod-shaped, non-motile, and covered
by a polysaccharide capsule [1], [2].
KLebsiella variicola
Scientific classification [3]:
Domain: Bacteria
Kingdom: Pseudomonadati
Phylum: Pseudomonadota
Class: Gammaproteobacteria
Order: Enterobacterales
Family: Enterobacteriaceae
Genus: Klebsiella
Species: K.variicola
K. variicola is a versatile bacterium capable of colonizing different hosts
such as plants, humans, insects and animals. Currently, K.variicola is gaining recognition as a cause of several human infections, especially
bloodstream infections (BSIs) [4].
The research will examine the key features of K. variicola’s genome and
proteome. Hopefully, the information studied will contribute to a more
complex understanding of K. variicola’s biology and, especially,
pathogenicity.
2 Methods
2.1 Information about genomic sequences, genome feature tables of K. variicola and other species considered it this research was obtained from
NCBI, genomic sequences of chromosome and plasmids from NCBI
Genome - GenBank assembly GCA_000019565.1. The genomic features
table of K. variicola is available for downloading on NCBI Genome
NCBI Genome . To obtain the features table the following steps were
executed: 1) Using Bash command “wget” the compressed gunzip file
“GCF_000019565.1_ASM1956v1_feature_table.txt.gz” was downloaded.
2) Using Bash command “gunzip” the received file was unpacked.
2.2 Standard information about the replicons, nucleotide composition of
every replicon in K. variicola was obtained using python tools (see
Supplementary materials/S1).
2.3 The counts of protein-coding genes and different RNA genes encoded
by each K. variicola replicon were compiled using Google Sheets (see
Supplementary materials/S2/sheet “per-replicons”). “COUNTIFS” function
was utilized, it counts the number of values satisfying defined criteria.
Furthermore, to determine the quantities of different rRNAs genes copies
the following Bash command was executed:
“cat *feature_table.txt | grep "^rRNA" | cut -f 14 | sort | uniq -c”, where
“*feature_table.txt” — the genomic features table of K. variicola (see
Methods/2.1).
To determine the number of tRNAs for each amino acid the following Bash
command was utilized:
“cat *feature_table.txt | grep "^tRNA" | cut -f 14 | sort | uniq -c”, where
“*feature_table.txt” — the genomic features table of K. variicola (see
Methods/2.1).
2.4 The inter-CDS gaps length diagram was constructed using Google
Sheets (see Supplementary materials/S2/sheet “interseq_lengths”).
Primarily, inter-CDS gaps lengths were calculated: for the first and last CDS
genes, the intergenic distance was calculated using the following formula:
D = L - EL + SF - 1, where D – distance length, L – chromosome
length, EL – last nucleotide position of last CDS (“end” column), SF – first
nucleotide position of the first CDS (“start” column).
For the remaining CDS, the gap length was derived using different formula:
D = Sn+1 - En - 1, where D – distance length, Sn+1 – first nucleotide
position of n+1 CDS, En – last position of n CDS.
Next, histogram lengths intervals were chosen and the chart was
generated. The “COUNTIFS” function was utilized to count the number of
values that satisfy the intervals.
2.5 To determine the position of the origin (oriC) of replication and
replication terminus (terC), GC-skew [5] and skewIT [6] methods were
utilized. To generate the GC-skew and skewIT charts Genskew_cc (a
commandline Genskew client) was used ("GitHub:
https://github.com/univieCUBE/genskew") . The client software was obtained
from its GitHub repository. All subsequent operations were performed in a
terminal of an IDE (specifically, in this research JetBrains PyCharm was
used). To access the usage instructions, the command «python3 -m
genskew -h» was run. After that, two analyses were performed using the
following commands on the sequence_chromosome.fasta file (K. variicola
chromosome sequence):
1. python3 -m genskew -s 2000 -w 2000 -o jpg -n1 G -n2 C -d 300 -of
/Users/user/Desktop ./sequence_chromosome.fasta — creates
GC-skew plot
2. python3 -m genskew -s 2000 -w 2000 -o jpg -n1 G -n2 C -d 300 -of
/Users/user/Desktop --skewIT true ./sequence_chromosome.fasta —
creates skewIT plot.
2.6 A comparison table featuring main genomic differences between
bacteria species within K. pneumoniae complex was made using python
script (see Supplementary materials/S1) and Google Sheets (see
Supplementary materials/S3/ sheet “CDS count”). CDS count was
calculated with Google Sheets “COUNTIFS” methods, G/C content and
chromosome length with the python script.
2.7 Protein length histogram was constructed in Google Sheets (see
Supplementary materials/S4/sheet “Prot_length_hist”). Primarily,
the lengths of every protein were found using following formula:
L = (E - S)/3 - 1, where L – protein length, E - last nucleotide position
of CDS, S – last nucleotide position of CDS.
Then, the maximum and minimum protein sizes were identified using
“MAX” and “MIN” functions respectively. Subsequently, the optimal bin
length was determined and the “COUNTIFS” function was used to calculate
the number of proteins contained within each bin. Next, the chart was
constructed.
3 RESULTS and DISCUSSION
3.1 Main genomic features
Primarily, it is crucial to state that K. variicola has 3 replicons, two
plasmids (pKP91 and pKP187) and a chromosome. The table below (Table
1) shows the standard information about the replicons:
Table 1. Standard replicon information.
Replicon
Size (bp)
GC content (%)
chromosome
5 641 239
57.29
pKP91
91 096
51.09
pKP187
187 922
47.15
Further researches gave more profound details (Table 2) about nucleotide
composition in K. variicola genome:
Table 2. Nucleotide composition of replicons.
Replicon
A
T
G
C
Chromosome
1203247
1206346
1612759
1618887
21.33%
21.38%
28.59%
28.7%
pKP91
22441
22112
23676
22867
24.63%
24.27%
25.99%
25.1%
pKP187
49794
49520
44794
43814
26.5%
26.35%
23.84%
23.31%
As the table shows, Chargaff’s rules apply for analyzed replicons [7].
3.2 Replicon composition
Furthermore, the amount of genes encoding proteins and different types of
RNA per every replicon was counted (Table 3):
Table 3. Number of Protein-Coding and RNA Genes per Replicon
Replicon
Chromosome
pKP91
pKP187
Cds with protein
5254
89
165
rRNA
25
0
0
tRNA
88
0
0
tmRNA
1
0
0
antisense RNA
2
0
0
ncRNA
11
0
0
pseudogene
108
12
25
It is obvious that the vast majority of genes responsible for ensuring vital
activity are encoded on the chromosome: A total of 5254 proteins are
encoded, 88 t-RNA genes, 25 rRNA genes and some minor types RNA
genes are present.
It is reasonable to assume that there are significantly more than 3 rRNA
genes, because they are assembled into clusters (is often referred to as
rDNA) of several copies encoding one of the three rRNAs: 8 copies of 16s,
8 copies of 23s and 9 copies of 5s. The rRNA genes form clusters
containing multiple copies in order to increase the synthesis of rRNA and,
eventually, ribosome formation [8].
Moreover, It should be noted that there are relatively many t-RNA genes in
the genome. Usually, their number does not exceed 61, since there are 61
possible coding codons in total (4
3
- 3 stop codons). But most organisms
possess significantly fewer than 61 distinct tRNAs. This is possible due to
the flexibility in codon-anticodon pairing described by the wobble rules. A
wobble base pair is a pairing between two nucleotides in RNA molecules
that does not follow Watson–Crick base pair rules. The four main wobble
base pairs are guanine-uracil (G–U), hypoxanthine–uracil (I–U),
hypoxanthine–adenine (I–A), and hypoxanthine–cytosine (I–C) [9].
The Bacterial Genetic Code (Translation Table 11) is characteristic to
K. variicola [10]. This translation table is identical to the Standard Genetic
Code [11] with one known exception: in some bacteria, such as E. coli and
Bacillus subtilis, the canonical stop codon “UGA” can encode tryptophan.
However, this “UGA-Trp” reassignment is not characteristic to K. variicola.
Table 4 presents the codon count per amino acid for translation table 11
and the tRNA gene count for each amino acid in K. variicola genome.
Table 4. Correspondence between the codon distribution of The Bacterial, Archaeal and Plant
Plastid Code and the tRNA gene repertoire in the K. variicola genome (a.a. – amino
acid, c.count – codon count, t.count – tRNA count).
a.a.
Ala
Arg
Asn
Asp
Cys
Glu
Gln
Gly
His
Ile
Leu
c.count
4
6
2
2
2
2
2
4
2
3
6
t.count
5
8
5
3
1
4
4
6
1
4
8
a.a.
Lys
Met
Phe
Pro
Ser
Thr
Trp
Tyr
Val
Sec
c.count
2
1
2
4
6
4
1
2
4
*
t.count
6
6
2
3
5
4
1
3
8
1
* Selenocysteine (Sec/U) is not encoded by the Standard Genetic Code and does not have a
dedicated codon.
As shown in Table 4, the number of tRNA genes in the genome exceeds
the number of corresponding codons for the majority of amino acids (13 out
of 20). The inverse is true for cysteine (Cys), histidine (His), proline (Pro),
and serine (Ser), where the codon count is greater than the tRNA gene
count. Tryptophan (Trp), threonine (Thr), and phenylalanine (Phe)
represent an equilibrium case, with equal numbers of codons and tRNA
genes.
Furthermore, analysis of the tRNA repertoire revealed the presence of a
selenocysteine-specific tRNA. This finding indicates that K. variicola utilizes
selenocysteine (Sec), suggesting that its proteome likely includes
selenoproteins [12].
There is a lack of information about plasmids pKP91 and pKP187 in
K. variicola. Here is information about plasmids pKP187 and pKP91 from
the related bacterium Klebsiella pneumoniae 342: pKP187 plasmid
encodes proteins involved in replication, partitioning/maintenance, arsenate
and tellurite resistance, also some regions of pKP187 consist of genes
taking part in carbohydrate metabolism. pKP91 plasmid encodes a putative
fusaric acid resistance gene as well as several oxidoreductase genes [13].
3.3 Inter-CDs distances
The intergenic distances between cds on the “+” strand were
calculated, and a histogram was constructed based on the obtained data
(Fig. 1.):
Fig. 1.The intergenic gaps length distribution on “+” strand.
It is important to clarify that the defined intergenic gaps contain no CDS on
the “+” strand but may contain features on the complementary strand,
including reverse-strand CDS and RNA genes. The proteome analysis in
section 3.6 indicates a high frequency of proteins with the length ≥50
amino acids. Consequently, many gaps exceeding 200 nucleotides in
length (calculated as: 50 codons * 3 nucleotides + 3 for a stop codon + ~50
for regulatory sequences) are likely to harbor CDS on the “-” strand.To
better assess genomic coding density, a histogram of intergenic gap
lengths (FIg. 2.) was constructed, accounting for coding sequences on both
the “+” and “-” strands:
Fig. 2. The intergenic gaps length distribution.
Analysis of the presented diagram demonstrates high-density gene
packing. The vast majority of intergenic intervals do not exceed 200–250
bp. Additionally, 807 instances of coding sequence (CDS) overlap were
detected. This pattern is consistent with and can be explained by the
operon-based organization of bacterial genomes [14].
Bacteria and archaea typically possess small genomes that are tightly
packed with protein-coding genes. The compactness of prokaryotic
genomes is commonly perceived as evidence of adaptive genome
streamlining caused by strong purifying selection in large microbial
populations. In such populations, even the small cost incurred by
nonfunctional DNA because of extra energy and time expenditure is
thought to be sufficient for this extra genetic material to be eliminated by
selection [15].
3.4 Localization of the origin and terminus of replication.
The positions of the putative origin and terminus of replication were
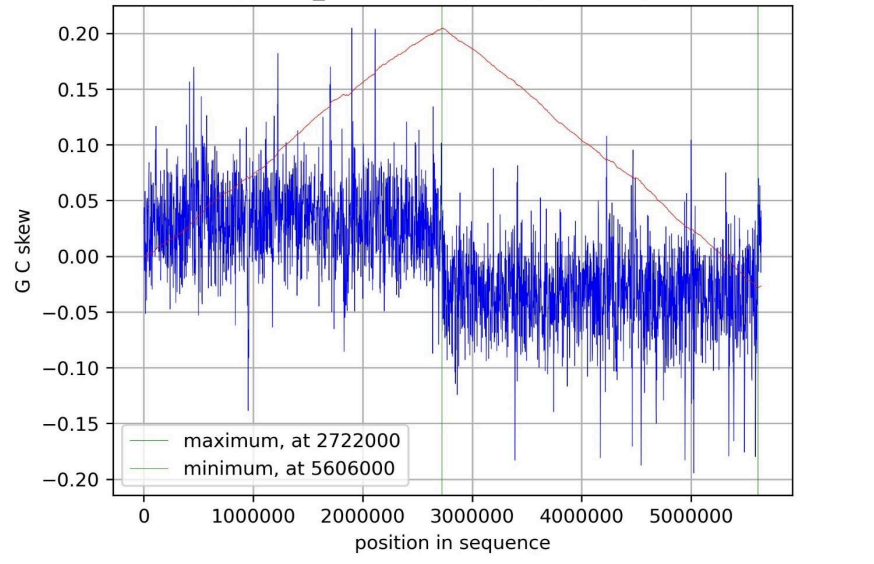
identified using the cumulative GC skew method (Fig. 3.):
Fig. 3. G/C-skew plot for K. variicola’s chromosome.
The cumulative GC skew analysis revealed a characteristic sawtooth
pattern across the circular chromosome of K.variicola. A global minimum in
the cumulative skew profile was identified at position 5606000 bp, marking
the predicted origin of replication (oriC). Conversely, a global maximum was
observed at approximately 2722000 bp, corresponding to the replication
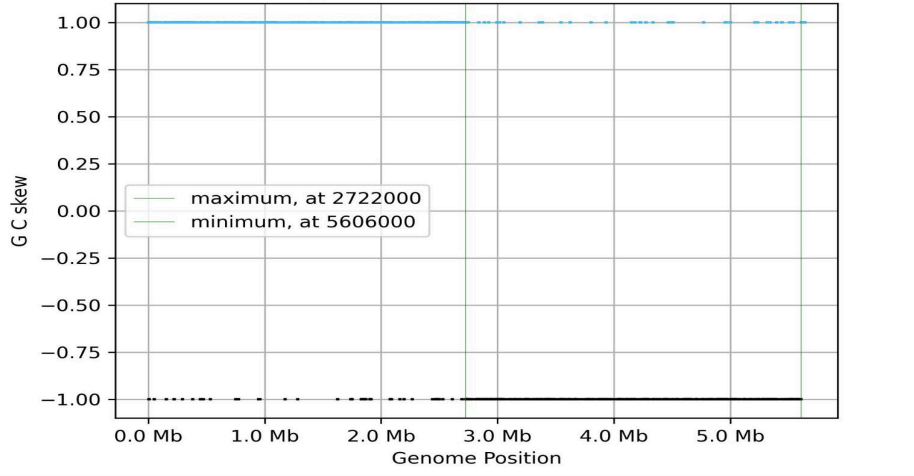
terminus (terC). The skewIT plot was also created (Fig. 4.):
Fig. 4. SkewIT plot for K. variicola’s chromosome.
The skewIT analysis identifies the window of maximal skewness, thereby
delineating the boundary between the A and B partitions. The position of
this boundary (2722000 bp) corresponds to the terminus of replication
(terC). And the window of minimal skewness indicates the approximate
location of the replication origin (oriC) near bp 5606000 (see Methods,
section 2.6). Thus, the skewIT analysis confirmed and refined the predicted
locations of oriC and terC.
3.5. Comparison of K. variicola genome with different
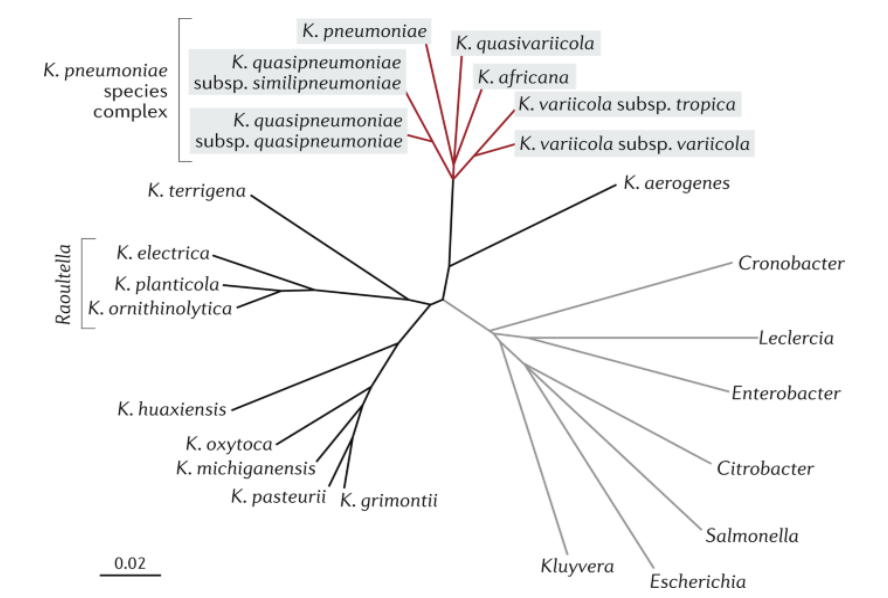
The Klebsiella pneumoniae complex (Fig. 5.) includes seven similar
species: K. pneumoniae subsp. pneumoniae, K. quasipneumoniae subsp.
quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, K.
quasivariicola, K. africana, K. variicola subsp. tropica and K. variicola
subsp. variicola (subsp. variicola is often referred to as “K. variicola” ) [16].
Fig. 5. Evolutionary tree of Enterobacteriaceae family [17].
The table below (Table 5) provides some information about K. pneumoniae
complex species chromosomes:
Table 5. Genomic features of K. pneumoniae complex species.
Species
Chromosome length (bp)
G/C content (%)
CDS count
K. variicola
5 641 239
57,29
5653
K. quasipneumoniae subsp. quasipneumoniae
5 343 479
58,14
5177
K. pneumoniae
5 333 941
57,48
5779
K. africana
5 243 981
57,16
5129
K. quasipneumoniae subsp. similipneumoniae
5 395 457
57,68
5507
K. variicola subsp. tropica
5 607 968
57,17
5632
K. quasivariicola
5 489 214
57,16
5396
Due to substantial overlap in their biochemical and phenotypic profiles,
these species cannot be reliably differentiated by traditional microbiological
methods. This taxonomic ambiguity, particularly the frequent
misidentification of K. variicola, has hindered research into its unique
biology and obscured its distinct clinical implications [17].
Significant attempts are now being made across the scientific community to
discover the best strategies for distinguishing between those species.
3.6. Protein profile
Fundamental research concerning the protein distribution based on their
length was conducted. The histogram below (Fig. 6.) illustrates the length
distribution of proteins found in K.variicola:
Fig. 6. Protein length distribution in K. variicola.
According to the diagram, the K.variicola’s proteome consists mostly of
relatively small proteins, approximately 75 percent of proteins have a length
of 0 - 400 amino acid residues.
Which refers to the fact that most of the proteins either structurally consist
of small amount of domains or exist as monomers, as it is usual in
prokaryotes [18].
4 Supplementary Materials
S1.Galiev_minireview.ipynb
— Python notebook, which was used to calculate replicon length, nucleotide composition, and G/C content.
S2.Minireview1
— Per replicons (list “per-replicons”) and interCDS distances (list “interseq_lengths”)
S3.KLebsiellas
— Information about Klebsiellas (Genomic feature tables of species)
[5] Antonio Marin, Xuhua Xia. GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: new substitution models incorporating strand bias. J Theor Biol. 2008;253(3):508-13
[6] Jennifer Lu, Steven L. Salzberg. SkewIT: The Skew Index Test for large-scale GC Skew analysis of bacterial genomes. PLoS Comput Biol. 2020;16(12):e1008439
[7] Keith L. Manchester. Historical Opinion: Erwin Chargaff and his ‘rules’ for the base composition of DNA: why did he fail to see the possibility of complementarity? Trends in Biochemical Sciences. 2008;33(2):65-70
[12] Yan Zhang, Jiao Jin, Biyan Huang, Huimin Ying, Jie He, Liang Jiang. Selenium Metabolism and Selenoproteins in Prokaryotes: A Bioinformatics Perspective. Biomolecules. 2022;12(7):917
[13] Derrick E. Fouts, Heather L. Tyler, Robert T. DeBoy, et al. Complete Genome Sequence of the N2-Fixing Broad Host Range Endophyte Klebsiella pneumoniae 342 and Virulence Predictions Verified in Mice. PLoS Genet. 2008;4(7):e1000141
[16] Ilse Verburg, Lucia Hernandez Leal, Karola Waar, et al. Klebsiella pneumoniae species complex: From wastewater to the environment. One Health. 2024;19:100880.