Практикум 12
или "ровнее, еще ровнее..."
Множественное выравнивание последовательностей белков
1. Сравнение выравниваний одних и тех же последовательностей разными программами
При выполнении задания были выбраны белки из семейства PF00998: B1PPP0_9HEPC; O93077_9HEPC; POLG_BVDVC; POLG_BVDVN.
Было сделано множественное выравнивание в Jalview, после чего использовалась программа Лизы Плешко для сравнения этих самых выравниваний. Большое спасибо ей за это :)
Ссылка на проект множественного выравнивания в Jalview
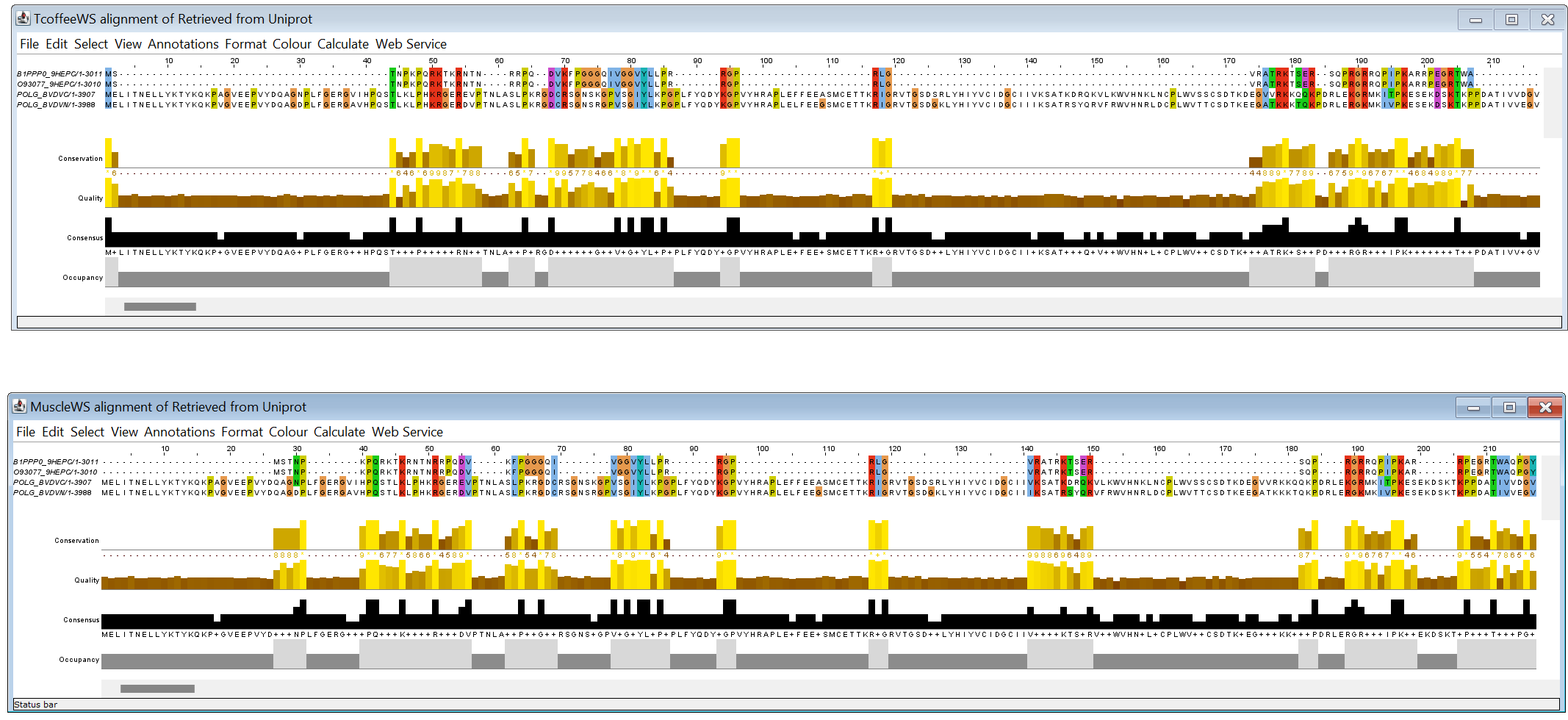
С помощью комманды "python compare_alig.py -a1 first_alignment.fasta -a2 second_alignment.fasta -o result_table.csv" было получено следующее:
Доля одинаково выравненных позиций в первом выравнивании: 0.44% (Tcoffee with defaults)
Доля одинаково выравненных позиций во втором выравнивании: 0.45% (Muscle with defaults)
Таблица одинаково выровненных позиций
Выравнивание Tcoffee with defaults в формате FASTA
Выравнивание Muscle with defaults в формате FASTA
Исходя из полученных данных, можно выделить несколько совпадающих крупных блоков, а также много мелких. Так, 78-140 позиции первого выравнивания соответствуют аналогичным 78-140 позициям второго, 485-664 первого соответствуют 458-641 второго, 1065-1182 первого и 1027-1144 второго. Это одни из наиболее крупных совпадающих блоков. Подобное разбиение соответствует разбиению выравниваний на гомологичные блоки. Сами блоки, при этом, выровнены одинаково, а короткие негомологичные промежутки между ними по-разному. Из всего сказанного можно придти к выводу о том, что гомологичные участки выравниваются разными программами одинаково, а выравнивание негомологичных участков может отличаться между собой.
2. Построение выравнивания по совмещению структур и сравнение его с выравниванием MSA
Из того же семейства PF00998 было выбрано 3 белка с PDB ID: 1YVF, 4KHR, 3HKW. Я построила выравнивание на сайте PDB и в Jalview. После их сравнения, пришла к выводу, что выравнивание в Jalview обладает большей точностью.

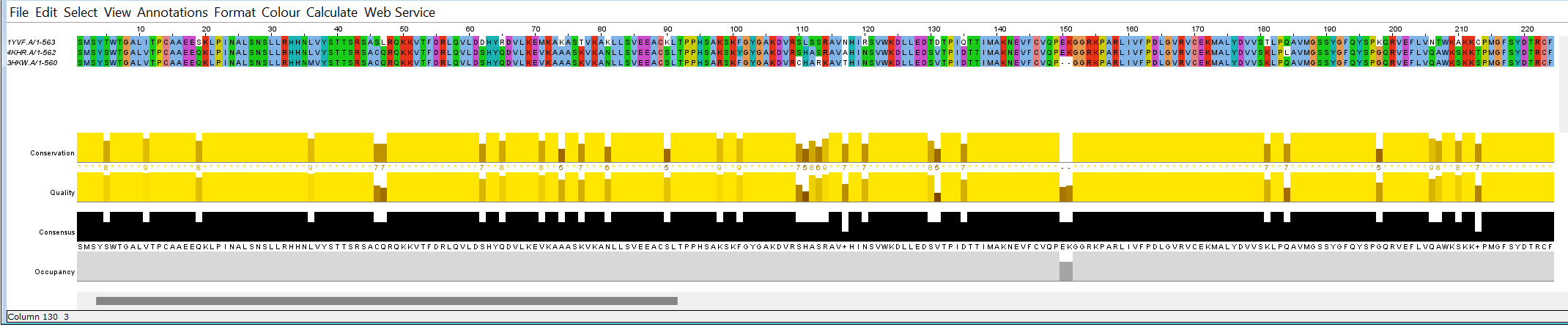
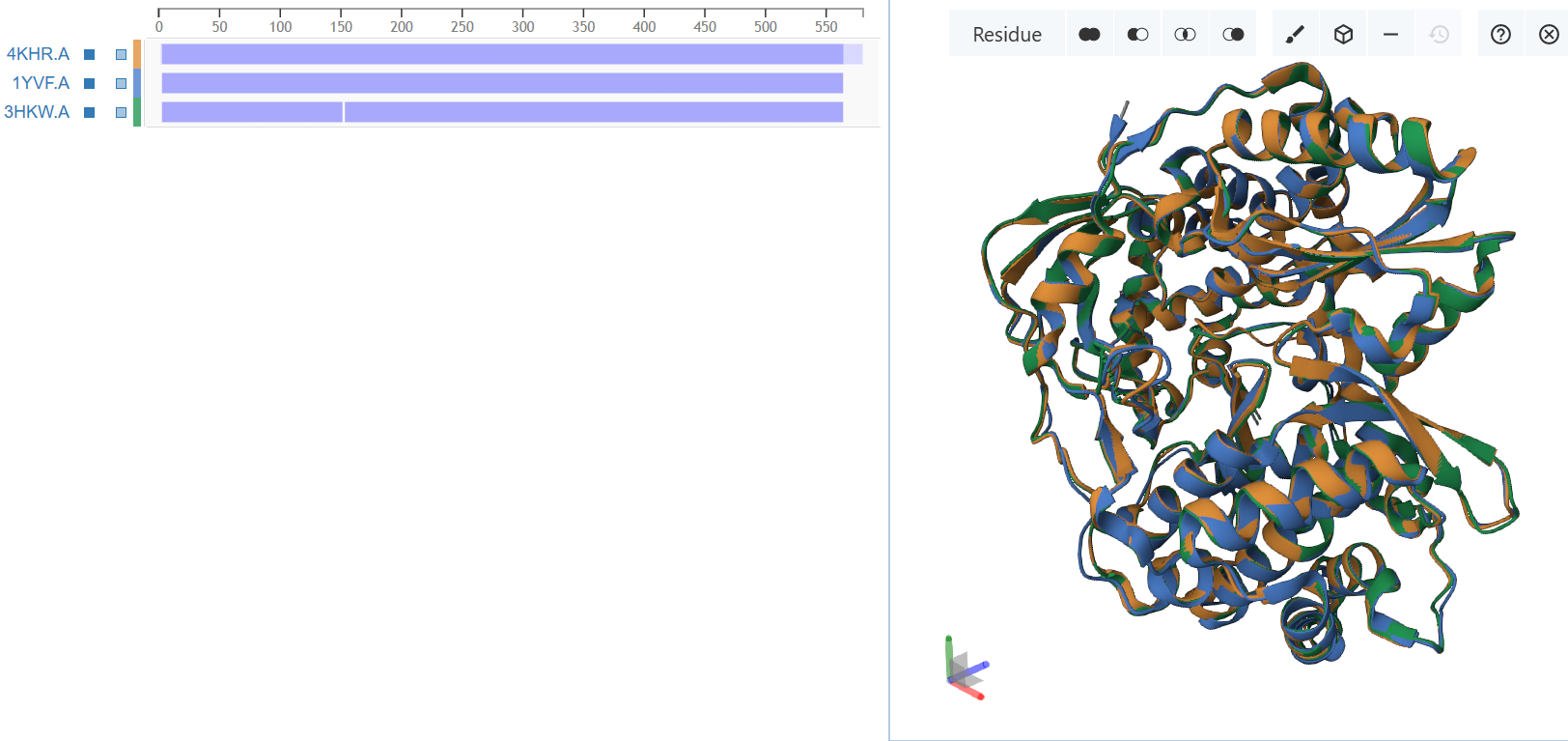
Все результаты и с сайта PDB, и с Jalview, а также пространственная структура (также с сайта PDB) подтверждают высокий уровень сходства последовательностей и наличие одинаковых вторичных структур.
Краткое описание одной из программ MSA
Множественное выравнивание последовательностей (англ. multiple sequence alignment, MSA) — выравнивание трёх и более биологических последовательностей, обычно белков, ДНК или РНК. В большинстве случаев предполагается, что входной набор последовательностей имеет эволюционную связь. Используя множественное выравнивание, можно оценить эволюционное происхождение последовательностей, проведя филогенетический анализ.
Множественное выравнивание последовательностей часто используется для оценки консервативности доменов белков, третичных и вторичных структур и даже отдельных аминокислотных остатков или нуклеотидов.
MUSCLE: MUltiple Sequence Comparison by Log-Expectation.
Сравнение множественных последовательностей с помощью логарифмического ожидания (MUSCLE) — это компьютерное программное обеспечение для выравнивания множественных последовательностей белковых и нуклеотидных последовательностей. Утверждается, что MUSCLE обеспечивает как лучшую среднюю точность, так и лучшую скорость, чем ClustalW2 или T-Coffee, в зависимости от выбранных параметров.
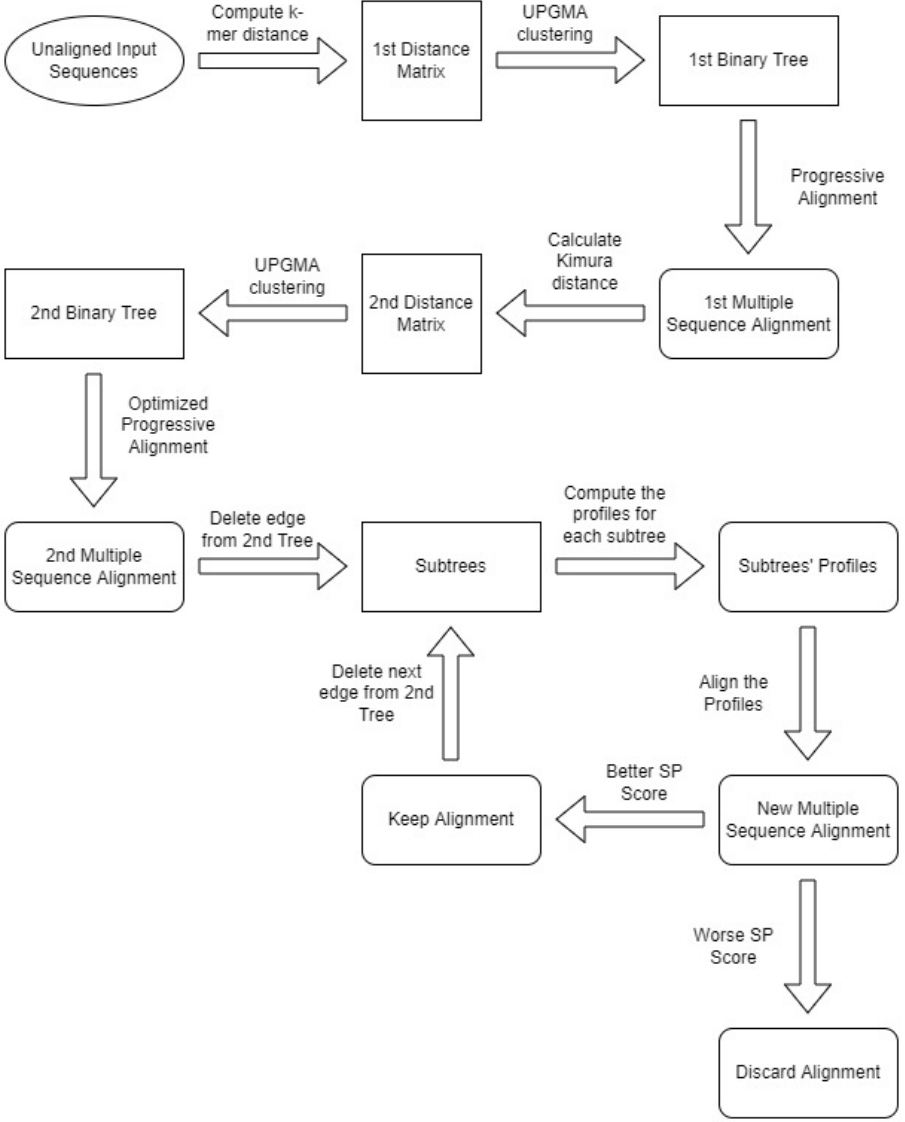
MUSCLE интегрирован в программное обеспечение DNASTAR Lasergene, Geneious и MacVector и доступен в Sequencher, MEGA, UGENE в качестве подключаемого модуля. MUSCLE также доступен в виде веб-сервиса через Европейскую лабораторию молекулярной биологии (EMBL) - Европейский институт биоинформатики (EBI). По состоянию на сентябрь 2016 года две статьи, описывающие MUSCLE, были процитированы в общей сложности более 19 000 раз.