Практикум 8
или "близнецы" blast
Нуклеотидный BLAST
Задание 1. Разные варианты BLAST для фрагмента ДНК
В качестве примера возьмем геном организма из предыдущего практикума, а именно геном прекрасной Lutra lutra. На примере фрагмента рассмотрим различия в работе разных алгоритмов BLAST.

Рис.1. Фотография чудесной Lutra lutra
Для выполнения практикума был выбран фрагмент хромосомы Y (accession NC_062297.1), с координатами 1755981-1761981, т.е. последовательность имеет длину 6000 нуклеотидов.
Ссылка на фрагмент в формате fasta
В данный фрагмент попали 3 гена. У одного из них есть аннотированная CDS, состоящая из 1 интрона. Полученный фрагмент с 3 генами можно увидеть на изображении ниже:

Рис.2. Схема расположения элементов(аннотированных) в выбранном фрагменте. Зеленый цвет - гены, фиолетовый - мРНК, красный - CDS.
В качестве семейства для поиска была выбрана группа Sauropsida (sauropsids), они же ящероподобные. Вполне далекие от выдрочек организмы:)
Результаты поиска при помощи разных алгоритмов
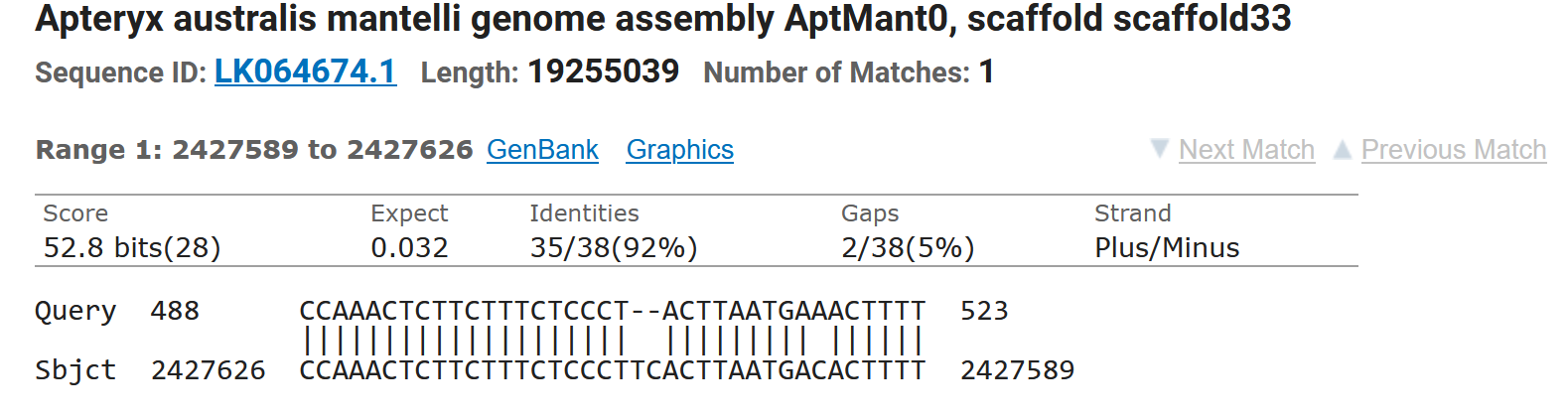
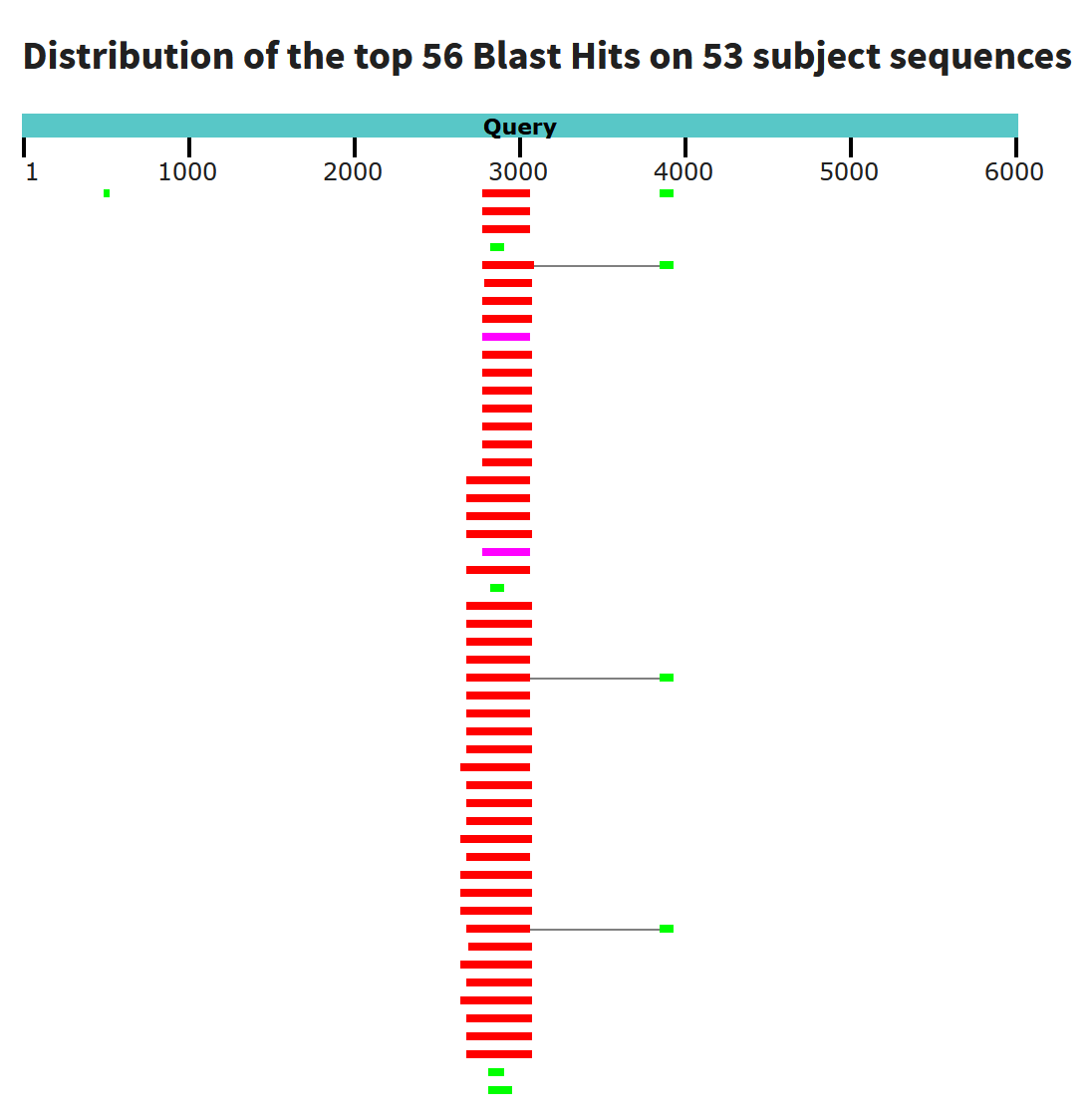
blastn: при использовании автоматических дефолтных параметров (просто указываем наши организмы) была всего 1 находка. Однако, уменьшая длину слова, находок получается больше (что логично, ведь при уменьшении длины слова увеличивается чувствительность поиска, хотя это и занимает больше времени). Итак, уменьшив длину слова до 16, мы получили 53 находки со средним покрытием 77-80%. Примечательно, что была одна находка с покрытием 92%, я решила посмотреть как же она выглядит графически, и вот что вышло:
Также можно посмотреть на графическое представление полученных результатов:
Ссылка на таблицу находок blastn
megablast: учитывая, насколько далеки друг от друга выдры и ящероподобные, логично, что алгоритм ничего не нашел:(
blastx: даже с дефолтной длиной слова алгоритм спокойно нашел 100 последовательностей, однако их покрытие составило примерно 70%. Небольшое наблюдение: все они hit shock factors, что в принципе, было ожидаемо. Что еще важно, так это, что домен определился в целом хорошо и что выравнивания красные, т.е. у них высокий score.
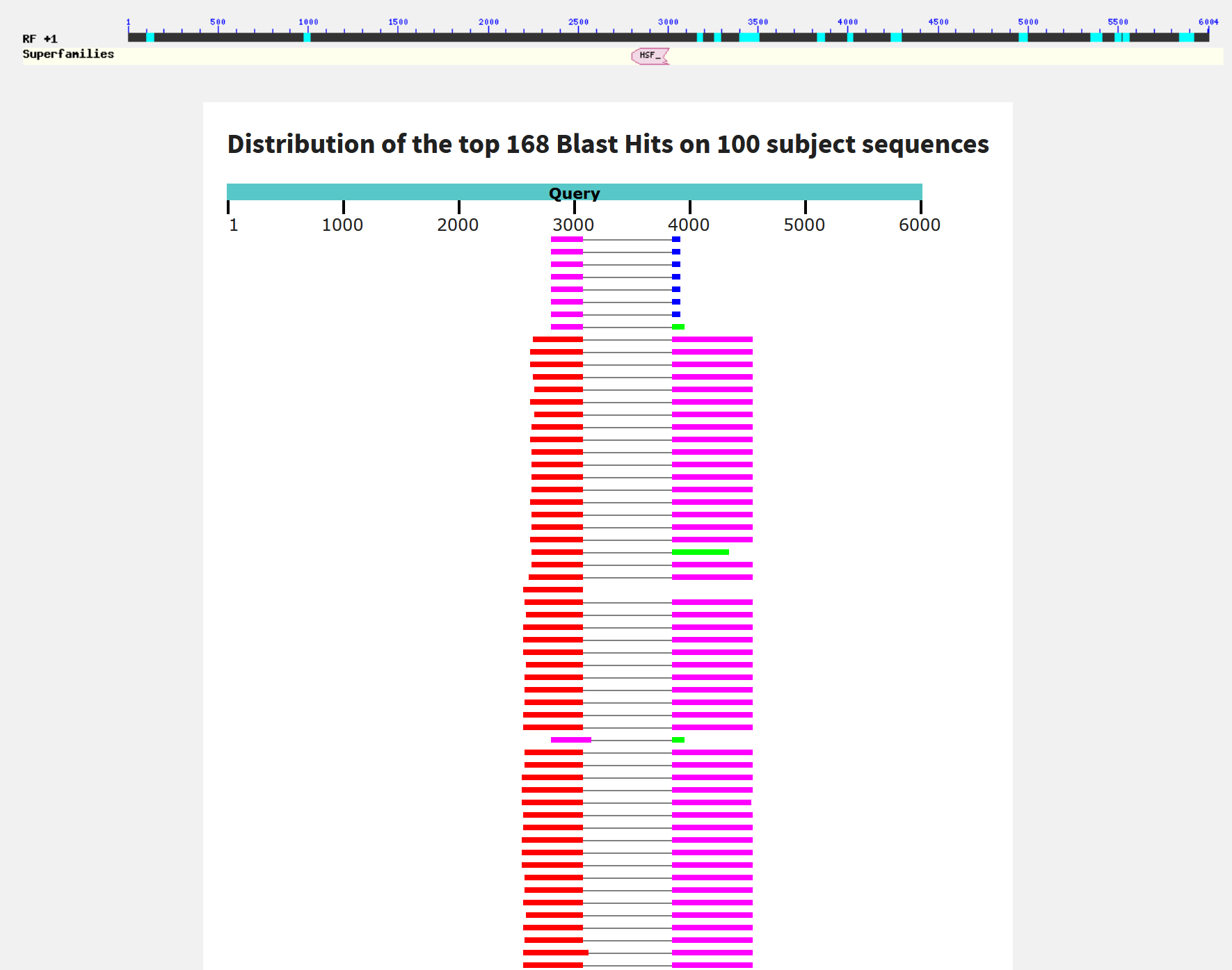
Ссылка на таблицу находок blastx
tblastx: оказалось, что данный алгоритм слишком затратен на таком объеме входных данных, а поменять базу данных сейчас мы не можем, поэтому и результатов нет:(
Особенности различных алгоритмов
blastn:определить принадлежность орагнизма к таксону, а также, чтобы сравнивать геномы не близкородственных организмов.
megablast:быстрый поиск среди очень похожих последовательностей.
blastx:определение кодирующих последовательностей.
tblastx:предсказание генов в последовательностях.
Поиск в геноме эукариота генов основных рибосомальных РНК по далекому гомологу
Ссылка на последовательность рРНК Escherichia coli
Чтобы BLAST работал, ему нужна специальная проиндексированная база данных. Что для этого было сделано? Ну, написаны скрипты:)
(А почему собственно нет?)
Ссылка на второй скрипт, который запустил необходимый нам поиск
Ссылка на третий скрипт, который собрал все результаты поиска вместе
В качестве запроса были взяты последовательности 16S и 23S рРНК E.coli. Для поиска использовался алгоритм blastn, т.к. эти РНК не кодируют белки, следовательно транслировать их не имеет смысла, ну и еще наши организмы далеко друг от друга по эволюции и тот же megablast, к примеру, ничего не найдет.
Ссылка для 16S, дополнительные данные
Ссылка для 23S, дополнительные данные
Итого было найдено 17 находок для 16S РНК и 13 находок для 23S РНК
Что можно заметить интересного еще? Так вот:
Если проводить поиск по каждой хромосоме отдельно, то получится 112 находок суммарно для 16S и 155 для 23S рРНК. При этом, e-value при поиске по отдельным хромосомам для тех находок, что пересекаются с поиском по всему геному сразу, на порядок меньше, чем при поиске по всему геному...Почему так?
Потому что при поиске по отдельной хромосоме статистически меньше вероятность нахождения похожего выравнивания, что и отражается в уменьшении e-value. Однако, как уже было замечено поиск по каждой отдельной хромосоме дает больше находок. Почему?
Снова из-за размеров баз данных, BLAST пропускает незначимые находки для каждой хромосомы и они копятся, а при поиске по всему геному такого не происходит.