Практикум 12. Сравнение выравниваний
Для сравнения двух выравниваний была написана программа на python.
Она последовательно принимает на вход пути к двум файлам с выравниваниями в формате FASTA (не должно быть пустых строк) и название файла для записи результатов (опционально).
Выдает файл с таблицей одинаково выравненных позиций и печатает в поток вывода долю одинаково выравненных позиций в каждом из выравниваний. Последовательности в двух выравниваниях могут быть по-разному отсортированы. В ходе работы программы они сортируются по ID.
У программы есть 3 аргумента: -a1, -a2, -o, отвечающие, соответственно за ввод названий файлов с первым выравниванием, вторым выравниванием и файлом, в который нужно записать результаты (по-умолчанию этот файл называется results-of_comparing.csv).
Пример вызова команды: "python compare_alig.py -a1 first_alignment.fasta -a2 second_alignment.fasta -o result_table.csv"
Программу можно скачать по ссылке.Работа программы была прверена на двух выравниваниях кутиназ.
Результаты сохранены в таблице.
Можно выделить 3 совпадающих блока: 92-109 позиции первого выравнивания соответствуют 109-119 позициям второго; 116-242 первого - 126-252 второго и 251-278 первого - 263-290 второго. Это разбиение соответсвует разбиению выравниваний на гомологичные блоки. Сами блоки выравнены одинаково, короткие негомологичные промежутки между ними - по-разному. Также различаются выравнивания N-конца, на котором последовательности тоже негомологичны (в одной из последовательностей произошла большая вставка, поэтому много гэпов).
Таким образом, одинаково выравниваются разными программами гомологичные участки, а выравнивание негомологичных может отличаться.
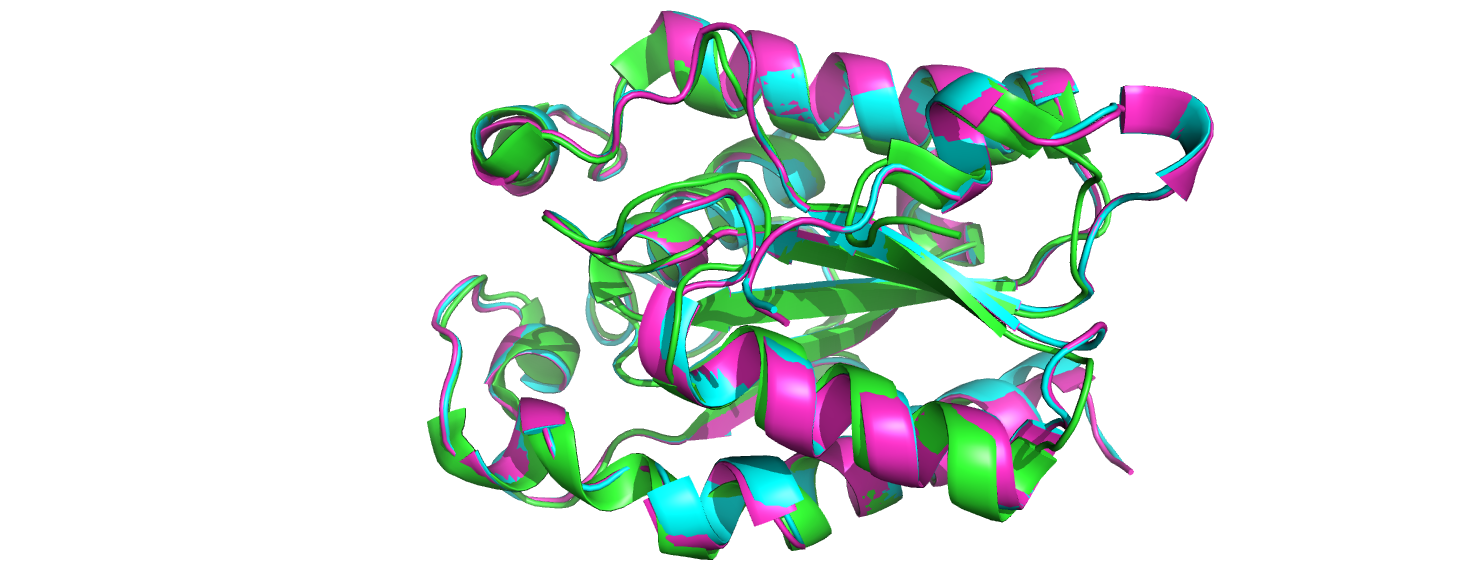
Было сделано выравнивание структур последовательностей трех кутиназ с PDB ID 3QPD, 1CUC и 1FFE. Выравнивание было сделано с помощью инструмента align в Pymol. Как видно из рисунка ниже, они выровнялись очень хорошо, струкуры почти полностью совпадают. Существенных различий нет: все стуктуры имеют одинаковую вторичную структуры.
Описание программы MAFFT
MAFFT (multiple alignment using fast Fourier transform) - алгоритм, основанный на прогрессивном выравнивании. Можно выделить несколько этапов:
1. Попарное выравнивание всех последоваетльностей. Особенностью алгоритма является использование Фурье-трансформации последовательности, при которой аминокислотные остатки заменяются их значениями объема и полярности. Это позволяет быстро обнаружить гомологичные участки при сравнении.
2. На основе результатов попарных выравниваний создается матрица расстояний между последовательностями. При этом используется особый способ подсчета расстояний, позволяющий уменьшить время работы программы и повысить точность.
3. Строится направляющее дерево, отражающее расстояние между последовательностями.
4. Прогрессивное выравнивание. Оно направлено от листьев дерева к корню. Сначала строится выравнивание наиболее близких последовательностей, после чего к нему добавляются все более отдаленные последовательности.
5. После этого процесс повторяется для увеличения точности. Вносятся корректировки в расположение гэпов и вставок.
В итоге удается быстро получить довольно точное множественное выравнивание.