Сборка генома
В ходе выполнения задания был собран геном бактерии Buchnera aphidicola str. Tuc7 и бактерии Kocuria carniphila, новый штамм которой секвенировали в моей лаборатории. На рисунке 1 показана электронная фотография кокурий и фотография старой чашки с разными штаммами.

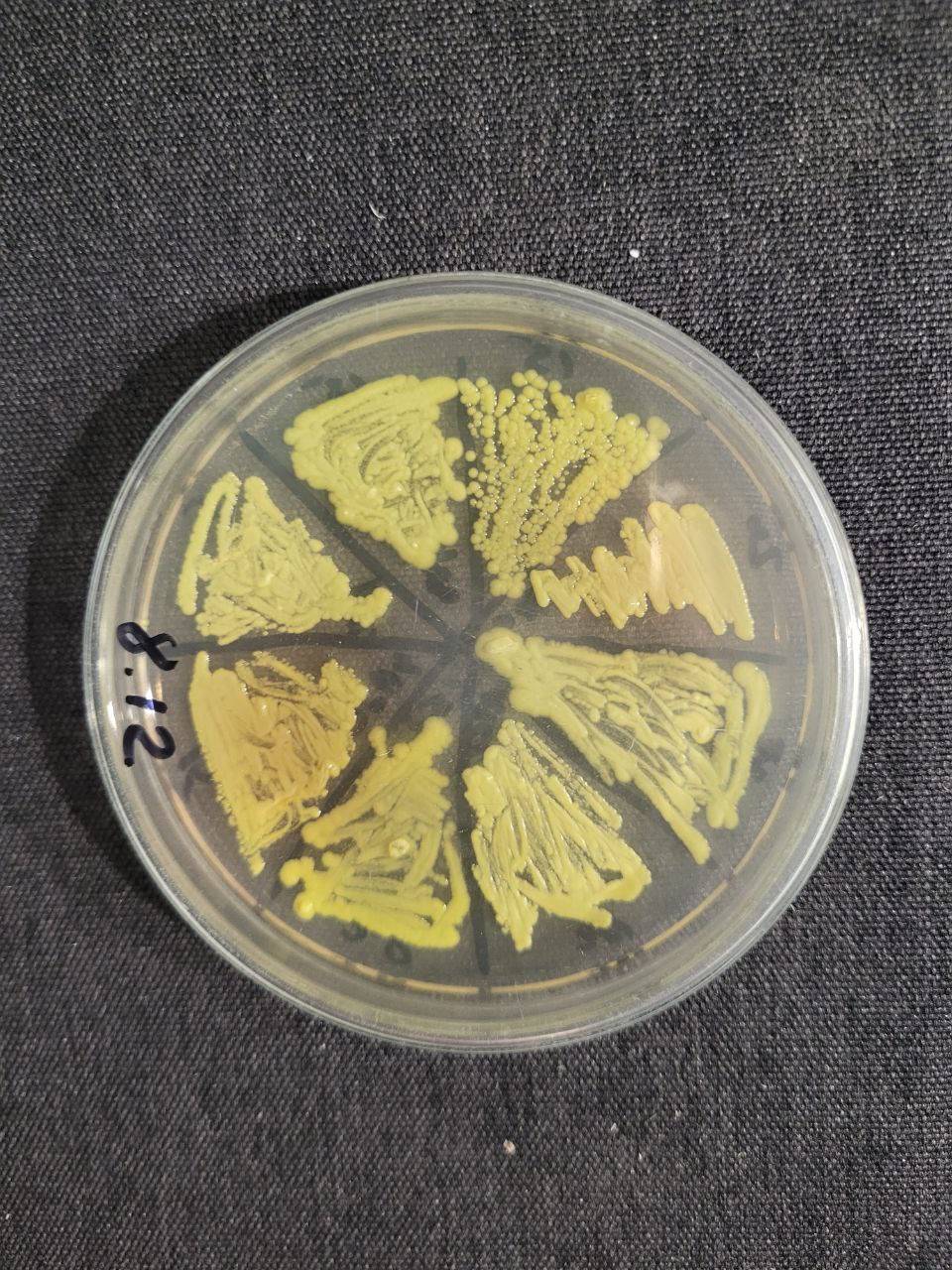
Рисунок 1. Электронная фотография кокурий и фотография чашки с разными штаммами кокурий.
Сборка Buchnera aphidicola
Сначала были скачаны результаты секвенирования ILLUMINA с помощью команды
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/006/SRR4240356/SRR4240356.fastq.gz
Затем из полученного файла были удалены прочтения с оставшимися адаптерами с использованием программы trimmomatic:
TrimmomaticSE -threads 15 -phred33 SRR4240356.fastq.gz trmmmed.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7
В результате было отброшено 153091 прочтений из 7511529 (2.04%).
Затем были удалены концевые нуклеотиды с качеством меньше 20, после чего были отброшены прочтения длиной менее 32 нуклеотидов. Для этого использовалась комманда:
TrimmomaticSE -threads 15 -phred33 trmmmed.fastq.gz final.fasta.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 TRAILING:20 MINLEN:32
В результате было удалено 305092 прочтений (4.15%).
Исходный файл весил 167 Мб. Файл, полученный после 1 запуска trimmomatic - 164 Мб, после 2 - 155 Мб.
Далее на основе отобранных прочтений были получены k-меры длин 31 с помощью команды:
velveth velvet 31 -short -fastq final.fasta.gz
Сборка была осуществлена с помощью программы velvetg:
velvetg velvet
Получилась сборка с N50 65554.
Сборка Kocuria carniphila
К файлам с прочтениями генома был приложен отчёт об их качестве. Оно достаточно высокое и адаптеров нигде не осталось, поэтому я не стала использовать Trimmomatic.
Сборка была осуществлена с помощью следующих команд:
velveth 988 31 -shortPaired -fastq.gz -separete wgs_S9505Nr1.1.fastq.gz wgs_S9505Nr1.2.fastq.gz
velvetg 988
Получилась сборка с N50 45 (полный провал).
Геном также был собран с помощью прогаммы SPAdes. Для этого была использована следующая команда:
spades.py -1 wgs_S9505Nr1.1.fastq.gz -2 wgs_S9505Nr1.2.fastq.gz -o 988_spades
Для обработки результатов, я написала скрипт на питоне (возможно, эти данные как-то выдавались программой, но я их не нашла):
input = open('988contigs.fasta', mode='r') #Сортировка файла по убыванию длин контигов
lst = input.read().split('>')
print(len(lst))
lst.sort(key=len, reverse=True)
output = open('988_contigs.fasta', 'w')
for elem in lst:
print('>'+elem, end = '', file=output)
names = list(map(lambda x: x.split('\n')[0], lst)) #Создания списка названий контигов
print(names)
contig1 = open('contig1.fasta', 'w') # Сохранение самых длинных контигов в отдельные файлы
contig2 = open('contig2.fasta', 'w')
contig3 = open('contig3.fasta', 'w')
print('>'+lst[0], file=contig1)
print('>'+lst[1], file=contig2)
print('>'+lst[2], file=contig3)
length = [] # Создание списков длин и покрытий
coverege = []
for name in names:
if len(name) > 1:
c = name.split('_')[-1]
l = name.split('_')[3]
length.append(int(l))
coverege.append(float(c))
print(length)
print(coverege)
total = sum(length) # Подсчёт N50
print(total)
n = 0
for l in length:
n += l
if n > total*0.5:
print(l)
break
for cov in coverege: # Поиск контигов с выбивающимися покрытиями
if cov > 1500 or cov < 60:
print(cov)
Получилось намного лучше, чем с помощью velvet. Всего 1306 контигов. N50 - 130097 нуклеотидов. Длины самых больших контигов - 317190, 260467, 235349 пар нуклеотидов. Их покрытие соответственно 286.317631, 324.443792, 365.61944. Общий размер генома - 3720398 нуклеотидов.
Контиги в среднем имеют покрытие около 300 (если не учитывать выбивающиеся). Контигов со слишком большим покрытием 3. Их покрытия равны 11724.989654, 1751.356932 и 1784.770915. Вторые два могут быть повторами, первый, скорее всего, - залёт. Также очень много контигов с покрытием меньше 2. Это тоже залёты. Все контиги с выбивающимися покрытиями имеют небольшую длину - в районе 300 нуклеотидов, это подтверждает залётную гипотезу. Видимо, я недостаточно аккуратно выделяла ДНК. Надеюсь, это не критично.
У моей бактерии нет полностью собранной хромосомы. Кроме того этот штамм значительно отличается по свойствам от других, вероятно, геномы тоже различаются. Поэтому я просто запустила поиск blastn с фильтрацией по таксону Kocuria и с параметрами, указаными на рисунке 2, для трёх самых длинных контигов. Для первого контига похожих последовательностей не было найдено. Для второго был найден фрагмент гена 16S рРНК кокурии MG733630.1 (Ссылка на файл с выравниванием). Для третьего контига нашёлся фрагмент генома кокурии OP296262.1 (Ссылка на файл с выравниванием), при этом выравнялось на очень небольшом участке. Что-то явно пошло не так.