Задание 1
Rho независимая терминация транскрипции у бактерий
Rho независимая терминация транскрипции осуществляется за счет образования шпильки на 3'-конце транскрипта. Эта шпилька содержит большое количество гуанина и цитозина, поэтому она очень прочная. За шпилькой следует участок, богатый урацилом, поэтому в том месте РНК и ДНК связываются между собой слабо. Шпильку узнает белок NusA, связанный с РНК-полимеразой. Из-за этого полимераза замедляется на участке, где много урацила. Это приводит к тому, что полимераза отваливается и транскрипция прекращается [1]. Этот сигнал должен обладать большой силой, т.к. иначе будет синтезироваться много лишней РНК. Это будет лишней тратой ресурсов и невыгодно для бактерий.
Сигналом Rho независимой терминации является G,C-богатая область, которая может образовывать шпильку сама с собой и за которой идет участок, богатый тимином. Для предсказания такимх сигналов можно использовать программу ARNold [2].
Для работы был взят геном бактерии Kocuria rhizophila (ASM456394v1). С помощью программы ARNold были найдены 171 терминатор транскрипции. Результаты доступны по ссылке. Например, был найден терминатор, который начинается на 66975 нуклеотиде скэфолда с идентификатором NZ_SPNK01000010.1. Его последовательность: TCAATAGTGTGTCTAGTTTGTTGATACTAGAaGTTTTATATTTT. Образующаяся шпилька показана на рисунке 1. Перед ним расположено несколько генов, учатсвующих в синтезе полисахаридов. Скорее всего, они образуют оперон, транскрипция которого заканчивается как раз найденным терминатором. Ближайший к нему ген - GlgB. Он кодирует белок, разветвляющий 1,4-альфа-глюкан. Этот ген заканчивается на 66,603 нуклеотиде. То есть между стоп-кодоном последнего гена в опероне и сигналом терминации транскрипции 372 нуклеотида.

Рисунок 1. Терминирующая шпилька одного из оперонов Kocuria rhizophila.
Источники:
1. Li K. et al. Escherichia coli transcription termination factor NusA: heat-induced oligomerization and chaperone activity //Scientific reports. – 2013. – Т. 3. – №. 1. – С. 2347.
2. Naville M. et al. ARNold: a web tool for the prediction of Rho-independent transcription terminators //RNA biology. – 2011. – Т. 8. – №. 1. – С. 11-13.
Задание 2
Я искала последовательности Шайна-Дальгарно в геноме Kocuria carniphila 988, который я сама собрала и проаннотировала в своей лаборатории. Про это написано здесь.
С помощью скрипта на питоне я вырезала 30 нуклеотидов перед старт-кодонами 100 генов и выравняла их в программе Jalview. Фрагмент результатов показан на рисунке 2. Они выглядят не очень консервативно. Я вырезала фрагмент выравнивания с 30 по 52 позиции. Из этих последовательностей 50 я взяла для обучения и 50 для тестирования. В качестве негативного контроля я взяла первые 22 нуклеотида первых 50 генов, т.к. внутри генов точно нет последовательностей Шайна-Дальгарно.

Рисунок 2. Выравнивание последовательностей перед старт-кодонами генов Kocuria carniphila 988.
Далее я использовала биопитон для построения PWM:
from Bio.Seq import Seq
from Bio import motifs
with open('study.txt', 'r') as fasta: #Получение списка последовательностей для обучения
gens = fasta.read().split('>')[1:]
instances = [Seq(gen.split('\n')[1]) for gen in gens if len(gen) > 1] #Преобразования последовательностей в специальный формат
m = motifs.create(instances) # Создание мотива
background = {"A": 0.4, "C": 0.6, "G": 0.6, "T": 0.4} #Учет частот нуклеотидов в геноме. У моей бактерии повышенный GC-состав, поэтому я выбрала такие значения.
pwm = m.counts.normalize(pseudocounts=background) #PWM
pssm = pwm.log_odds(background) #Position-Specific Scoring Matrices
with open('test.txt', 'r') as fasta: #Получение списка последовательностей для тестирование
gens = fasta.read().split('>')[1:]
test_weights = []
test = [Seq(gen.split('\n')[1]) for gen in gens if len(gen) > 1] #Преобразования последовательностей в специальный формат
for seq in test:
a = pssm.calculate(seq) #Рассчет расстояний для всех последовательностей
test_weights.append(a)
with open('negative.txt', 'r') as fasta: #То же самое для негативного контроля и материала для обучения
gens = fasta.read().split('>')[1:]
negative_weights = []
negative = [Seq(gen.split('\n')[1]) for gen in gens if len(gen) > 1]
for seq in negative:
a = pssm.calculate(seq)
negative_weights.append(a) study_weights = [] for seq in instances: a = pssm.calculate(seq) study_weights.append(a)
print(pwm.consensus)
#Консенсусdistribution = pssm.distribution(background=background, precision=10**4) #Рассчет порога веса
threshold = distribution.threshold_fpr(0.01)
print("%5.3f" % threshold)
Полученная PWM выглядит вот так:

Консенсус: CCCACCGCCCCCCGGGGAGGGC
Далее я построила LOGO с помощью скрипта на питоне, который я взяла отсюда. Результат на рисунке 3.
Рисунок 3. LOGO участка, где должна быть последовательность Шайна-Дальгарно.
Порог веса равен 3.981. Вероятность случайно сгенерировать последовательность (с учетом встречаемости нуклеотидов) с таким или большим весом равна 0.01. На рисунке 4 показаны распределения весов последовательностей из разных групп. Видно, что очень небольшая доля последовательностей из негативного контроля имеет веса, превышающие пороговый.
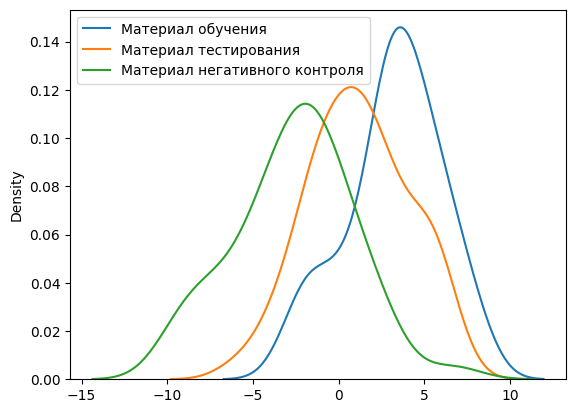
Рисунок 4. Распределение весов последовательностей.
У всего этого может быть практическое применение: можно предсказывать, транслируются ли открытые рамки считывания. Это может быть полезно в изучении гипотетических белков. Однако этот метод предсказания не очень точен, т.к. последовательность Шайна-Дальгарно неконсервативна несмотря на то, что участок 16s рРНК, который с ней связывается (anti-SD), очень консервативен. же давно известно, что рибосома может находить нужные старт-кодоны даже без anti-SD, т.к. есть и другие механизмы регуляции (Melancon P. et al. The anti-Shine-Dalgarno region in Escherichia coli 16S ribosomal RNA is not essential for the correct selection of translational starts //Biochemistry. – 1990. – Т. 29. – №. 13. – С. 3402-3407).