Цель занятия используя пакет модулей RDkit предложить аналог ибупрофена :
-на сайте PubChem найти все радикалы c азидом для Click Chemistry и скачать их SMILES нотации
-Найти формулу ибупрофена и предложить способ изменения его SMILES для эмуляции продукта Click Chemistry
-Заменить в найденых радикалах азидную группу на модифцированный ибупрофен.
-Превратить новые SMILES в объекты-молекулы
-Отобрать те молекулы, которые удовлетворяют правилу пяти Lepinsky
Загрузим модули RDkit
In [9]:
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
import numpy as np
from IPython.display import display,Image
Нарисуем ибупрофен и его производные для Click Chemistry
In [10]:
ibu=Chem.MolFromSmiles('CC(C)CC1=CC=C(C=C1)C(C)C(=O)O')
AllChem.Compute2DCoords(ibu)
display(ibu)
#Замена изопропила -C#C
modif_ibu = Chem.MolFromSmiles('C#CC1=CC=C(C=C1)C(C)C(=O)O')
AllChem.Compute2DCoords(modif_ibu)
display(modif_ibu)
#добавление кольца
template = Chem.MolFromSmiles('N1C=C(N=N1)C1=CC=C(C=C1)C(C)C(=O)O')
AllChem.Compute2DCoords(template)
display(template)



Посчитаем параметры для правила Лепински
In [11]:
import rdkit.Chem.Lipinski as Lipinksy
print Lipinksy.NumHDonors(ibu)
print Lipinksy.NumHAcceptors(ibu)
print Lipinksy.rdMolDescriptors.CalcExactMolWt(ibu)
print Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0]
Загрузим скаченные SMILES азидов и отфильтруем
In [12]:
strings=np.genfromtxt('3431467748381165471.txt',dtype=np.str)
smiles = []
for line in strings:
if len(line[1]) < 30 and not '.' in line[1]:
smiles.append(line[1])
Построим новые молекулы и отфильтруем
In [13]:
#for smi in smiles[:1500]:
good_new_smiles=[]
N=0
#all_mol = len(smiles)
all_mol = 100
min = Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0]
min_index = 0
#Новую молекулу лучше создавать в try из-за возможных битых Smiles
for smi in smiles[:all_mol]:
if 'N=[N+]=[N-]' in smi:
newsmi=smi.replace('N=[N+]=[N-]','N1C=C(N=N1)C1=CC=C(C=C1)C(C)C(=O)O') #replace with template
else:
continue
try:
newmol=Chem.MolFromSmiles(newsmi)
if Lipinksy.NumHDonors(newmol) <= 5 and Lipinksy.NumHAcceptors(newmol) <= 10 and Lipinksy.rdMolDescriptors.CalcExactMolWt(newmol) <=500 and Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(newmol)[0] <= Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0]:
good_new_smiles.append(newmol)
AllChem.Compute2DCoords(newmol)
display(newmol)
print N
N=N+1
if Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(newmol)[0] < min:
min = Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(newmol)[0]
min_index = N-1
except:
pass
print 'good molecules: ', float(N)/float(all_mol)*100,'%'
print 'molecule with minimal log P: %i with number %i' %(min,min_index)
display(good_new_smiles[min_index])























































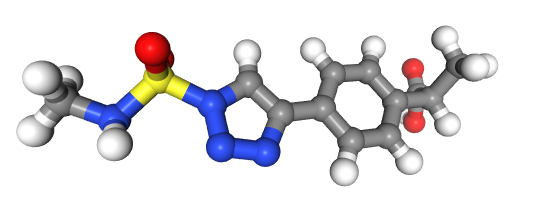
Пострим 3D модель для 12 соединения, так как это считается самой хорошей молекулой.
In [14]:
m3d=Chem.AddHs(good_new_smiles[12])
Chem.AllChem.EmbedMolecule(m3d)
AllChem.MMFFOptimizeMolecule(m3d,maxIters=500,nonBondedThresh=200)
Out[14]:
In [15]:
import nglview as nv
nv.show_rdkit(m3d)