Молекулярная динамика биологических молекул в GROMACS
Цель данного занятия ознакомится с возможностями моделирования молекулярной динамики. Предлагалось 4 различных систем для моделирования. Была выбрана система Моделирование самосборки липидного бислоя.
Моделирование самосборки липидного бислоя
Исходные файлы:
- дополнительной топологии для липида DPPC.
- параметры для липидов.
- координаты одного липида.
- Файл-заготовка тополгии системы.
- файл праметров для минимизации энергии.
- файл праметров для "утряски" воды.
- файл праметров для молекулярной динамики.
На основе одного липида создали ячейку с 64 липидами.
!genconf -f dppc.gro -o b_64.gro -nbox 4 4 4
С помощью editconf преобразовали dppc.gro и b_64.gro в pdb файлы.
!editconf -f dppc.gro -o dppc.pdb
!editconf -f b_64.gro -o b_64.pdb
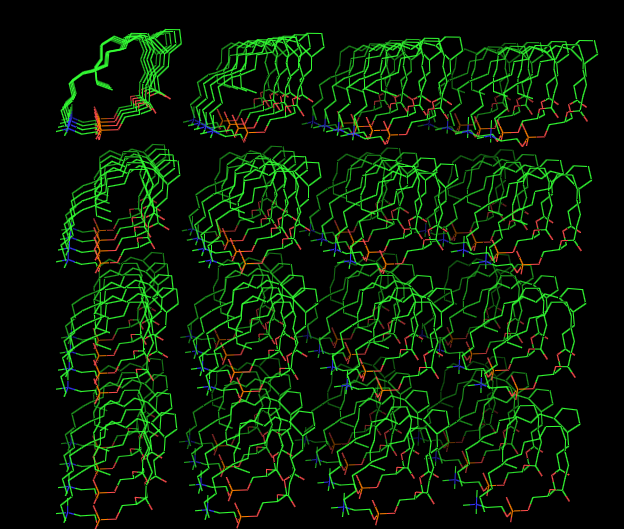
В файле b.top нашли правильное количество липидов: DPPC 64.
Сделали небольшой отступ в ячейке от липидов, что бы добавить примерно 2500 молекул воды.
!editconf -f b_64.gro -o b_ec -d 0.5
Провели оптимизацию геометрии системы, что бы удалить "плохие" контакты молекул.
!grompp -f em -c b_ec -p b -o b_em -maxwarn 2
!mdrun -deffnm b_em -v
Посмотрели изменение максимальной силы в ходе оптимизации геометрии. Начальное и конечное значение максимальной силы - 4.37970e+05 и 6.1937860e+02. Сила заметно уменьшилась.
Добавили в ячейку молекулы воды типа spc.
!genbox -cp b_em -p b -cs spc216 -o b_s
Провели "утряску" воды:
!grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
!mdrun -deffnm b_pr -v
Переформатировали b_pr.gro и b_s.gro в pdb формат.
!editconf -f b_pr.gro -o b_pr.pdb
!editconf -f b_s.gro -o b_s.pdb
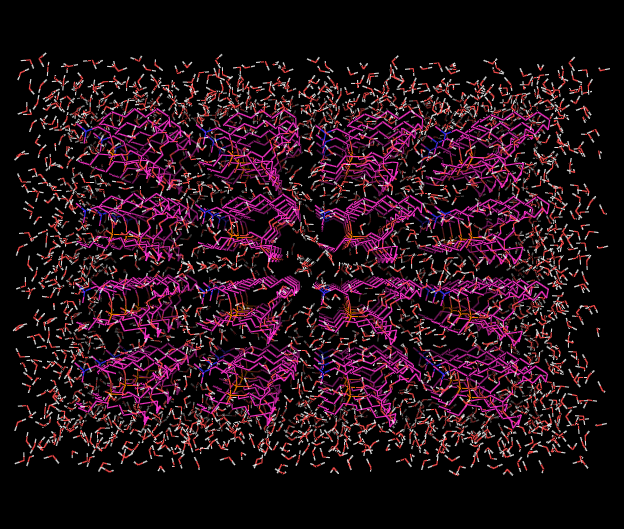
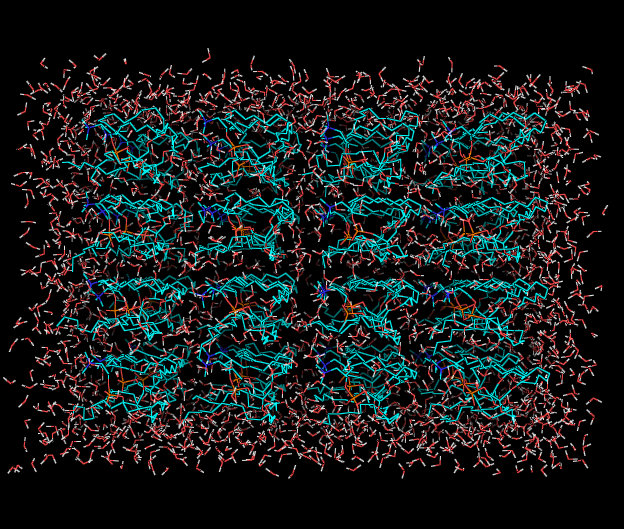
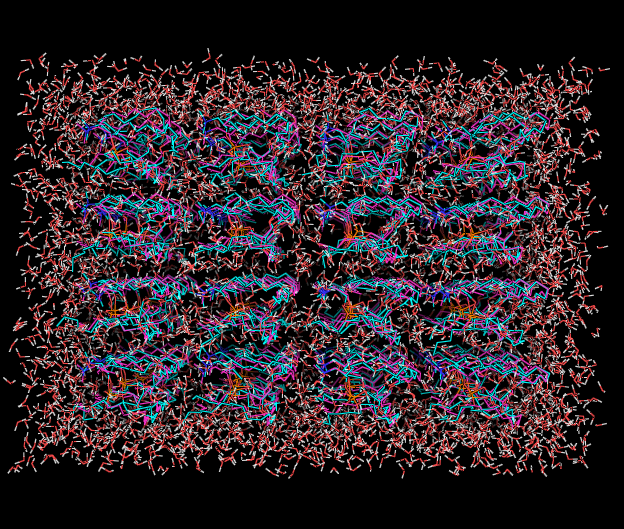
Видно, что конформации сильно различаются.
Запустили основное моделирование на суперкомпьтере - последний этап практикума.
!sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log -t 350 -p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm b_md -v