Работа производилась с человеческой хромосомой 12. Работа выполнялась для чтения экзома пациента, которые нужно картировать на участок определённой хромосомы человека сборки генома hg19 (хромосомы 12 в моём случае), которые находятся в файле с одноконцевыми чтениями chr12.fastq).
С помощью программы FastQC (команда fastqc chr12.fastq) был получен архив и файл html, содержащие информацию о качестве прочтений.
В числе прочего программа FastQC создала картинку, визуализирующую качество прочтения. По горизонтали снизу числами обозначены позиции в ридах, а по вертикали отложено качество рида. Видно, что качество прочтения сильно хуже в концах прочтения; с этим мы уже сталкивались при работе с хроматограммами.
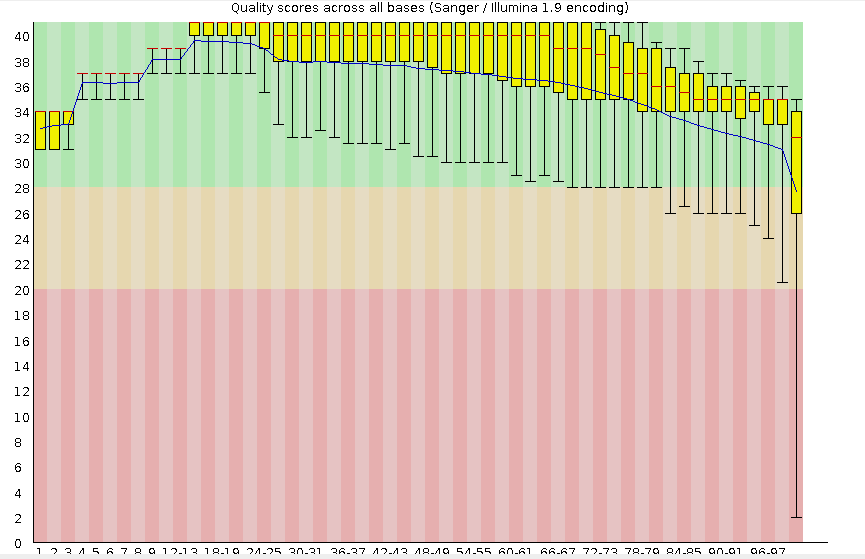
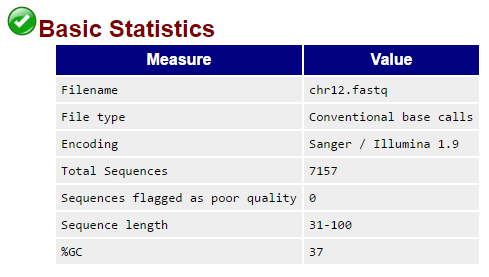
Кроме того, в таблице Basic statistics собраны основные данные о прочтении: мы узнаем конкретную версию секвенатора Illumina (Illumina 1.9), общее число чтений (7157) и диапазон длины ридов (31-100), а также GC-состав прочтений (37 %).
Далее производилась очистка чтений с помощью команды Trimmomatic. Я запускала данную программу через сервер kodomo с опциями TRAILING:20 (отрезает с конца каждого чтения нуклеотиды с качеством ниже 20) и MINLEN:50 (удаляет риды короче 50 нуклеотидов):
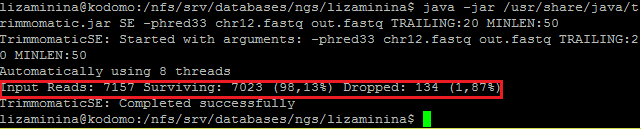
Красной рамкой обведена выданная программой информация. Из неё видно, что из 7157 чтений после чистки осталось 7023 (98,13 %), а 134 (1,87 %) было отброшено.
Чтобы посмотреть, какие риды были отброшены, я запустила программу FastQC с файлом, содержащим очищенные последовательности (out.fastq). Результатом выполнения программы является файл html и архив.
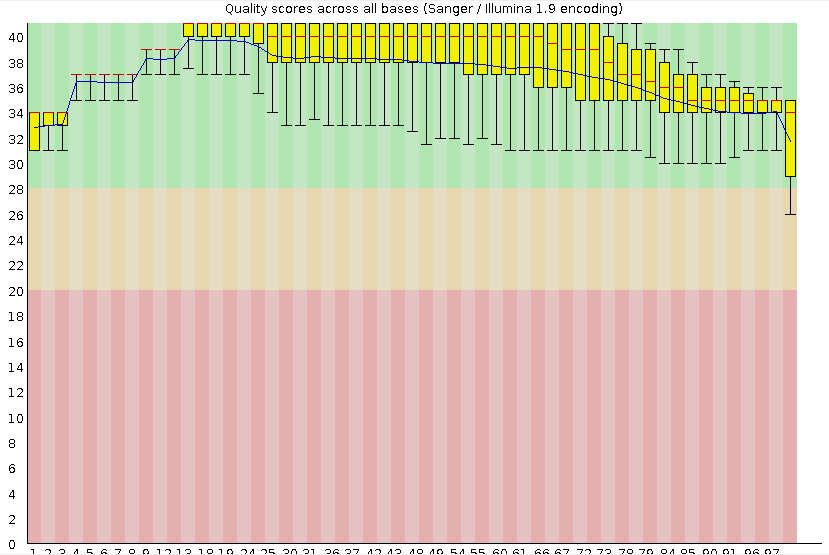
Как видно из схемы, качество прочтений существенно выше, разница в качестве между серединой и концами прочтений сглажена.
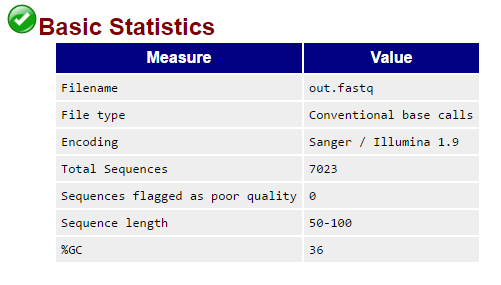
О повышении качества прочтений свидетельствует и таблица Basic statistics: количество ридов уменьшилось (7023 против 7157 до очистки) из-за отбраковки совсем плохих и коротких (программа отбросила риды длиной до 30 нуклеотидов), длина ридов от 50 до 100 нуклеотидов. Таким образом, программа Trimmomatic позволила повысить качество прочтений и отбросить совсем короткие риды.
Далее с помощью программы BWA надо было откартировать очищенные чтения. Cначала я проиндексировала референсную последовательность хромосомы 12 при помощи команды bwa index chr12.fasta:
Затем я построила выравнивание прочтений и референса в формате .sam с помощью команды, приведённой на рис.:
Анализ выравнивания был произведён при помощи пакета программ samtools. выравнивание чтений с референсом в бинарный формат .bam, запустив команду samtools view с опциями -b (выходной файл бинарный) и -о (указание имени выходного файла): samtools view -b -o aln.bam aln.sam. Потом я отсортировала выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения. Это было сделано с помощью команды samtools sort -T/tmp/aln.sorted -o aln.sorted.bam aln.bam. После этого я проиндексировала отсортированный .bam файл с помощью команды samtools index aln.sorted.bam. Наконец я получила информацию о том, сколько чтений откартировалось на геном, используя команду samtools idxstats aln.sorted.bam. Последовательность команд и конечный результат представлены на рисунке ниже:
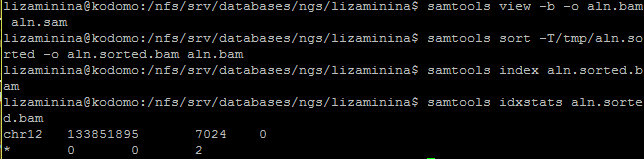
Программа выдает информацию о картировании в следующей последовательности: название последовательности, длина последовательности, число картировавшихся ридов, число некартировавшихся ридов. Таким образом, в моем случае картировалось 7024 рида (что странно, так как всего после чистки осталось 7023 рида), и не было некартировавшихся ридов.
При помощи команды samtools mpileup с опциями -uf (определяют формат входных и выходных файлов) для получения файла .bcf с полиморфизмами. Различия между референсом и чтениями были определены с помощью команды bcftools call с опциями -cv пакета bcftools, результатом работы которой является файл в формате .vcf. Синтаксис команд см. на рисунке ниже.
Всего было найдено 3 делеции и 35 замен. В таблице ниже приведены данные по трём полиморфизмам.
| Координата | Тип полиморфизма | В референсе (было) | В чтении (стало) | Глубина покрытия данного места | Качество чтений в данном месте |
| 9822387 | Замена | C | G | 31 | 190.009 |
| 9848454 | Замена | A | C | 48 | 198.009 |
| 9849138 | Делеция | ataaagtaaa | ataaa | 2 | 179.468 |
Качество покрытия во всех трёх случаях неплохое, а вот глубина в случае делеции сильно меньше, чем у двух замен.
Далее нужно было проаннотировать только полученные snp помощью программы annovar по следующим базам данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar. Для аннотации файла с snp с помощью предложенных баз данных используйте скрипт annotate_variation.pl. Программа annovar работает с файлами в формате avinput. Получить файл в таком формате можно при помощи скрипта convert2annovar.pl. На рисунке ниже показано, как он был запущен.
После этого было необходимо определить, какие из найденных замен имеют rs (идентификатор в базе данных однонуклеотидных полиморфизмов, то есть они уже изучены или хотя бы известны). На рисунке ниже показано, какая команда была выполнена и что было выдано в результате:
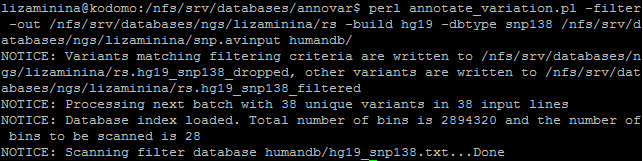
Таким образом, 2894320 SNP имеют rs.
Аннотация SNP по базе данных refgene была запущена следующей командой:
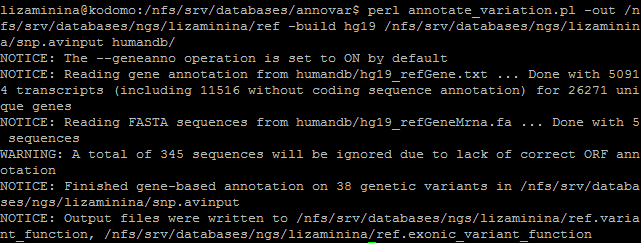
SNP делятся на типы het и hom. В итоге было найдено 14 SNP типа het и 24 SNP типа hom. Из них в экзоны попало 3 SNP, в 3'-нетранслируемую область - 9 замен, а остальные попали в интроны, причём один SNP затронула интрон некодирующей РНК (ncRNA). SNP затронули четыре гена:
1) CLEC2D кодирует клеточный рецептор натуральных киллеров, принадлежащий к С-типу лектинового семейства.
2) RPSAP52 - псевдоген 52 рибосомного белка SA.
3) HMGA2 - негистоновый хромосомный белок группы HGM.
4) TMEM263 - трансмембранный белок.
В таблице ниже приведена информация об аминокислотных заменах, связанных с SNP:
| Ген | Позиция | Референс | Чтение | Аминокислотная замена |
| CLEC2D | 9822387 | C | G | N19K |
| CLEC2D | 9833524 | C | G | L23V |
| CLEC2D | 9833628 | C | T | S57S |
Аннотация SNP по базе данных dbsnp была запущена следующей командой:
Аннотация по базе dbsnp показала, что 28 замен имеют rs, а 10 замен не имеют.
Аннотация SNP по базе данных 1000 genomes была запущена следующей командой:
Аннотация по базе 1000 genomes позволила определить, что наиболее популярной заменой является замена Т на С, а частоты замен варьируют от 0,0108 до 0,7045.
Аннотация SNP по базе данных GWAS была запущена следующей командой:
Из этой базы данных была взята клиническая аннотация SNP: обнаруженные SNP могут быть связаны с развитием диабета 1 типа, другие влияют на структуру мозга, рост и плотность костного вещества.
Аннотация SNP по базе данных Clinvar была запущена следующей командой:
Полностью ознакомиться с результатами аннотации по разным базам данных можно в файле Excel.
В таблице ниже перечислены команды, использованные при выполнении практикума:
| Команда | Что делает |
| fastqc chr12.fastq | Анализ качества ридов |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr12.fastq TRAILINF:20 MINLEN:50 | Очистка чтений (Trimmomatic); TRAILING удаляет с конца чтения нуклеотиды с качеством менее 20, MINLEN убирает риды с длиной меньше 30 |
| bwa index chr12.fasta | Индексирование референсной последовательности |
| bwa mem chr12.fasta out.fastq > aln.sam | Построение выравнивания прочтений и референса в формате .sam |
| samtools view -bh -o aln.bam aln.sma | Переводит выравнивание в бинарный формат .bam |
| samtools sort -T/tmp/aln.sorted -o aln.sorted.bam aln.bam | Сортировка выравнивания по координате референса в начале чтения |
| samtools index aln.sorted.bam | Индексирование отсортированного файла в формате .bam |
| samtools idxstats aln.sorted.bam | Информация о количестве чтений, откартированных на геном |
| samtools mpileup -uf chr12.fasta aln.sorted.bam > snp.bcf | Получение файла с полиморфизмами в формате .bcf |
| bcftools call -cv snp.bcf > snp.vcf | Получение файла со списком отличий между референсом и чтениями в формате .vcf |
| perl convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/lizaminina/snp.vcf > /nsf/srv/databases/ngs/lizaminina/snp.avinput | Перевод файла в формате .vcf в формат avinput, с которым работает программа annover |
| annotate_variation.pl -filter -out /nfs/srv/databases/ngs/lizaminina/rs -build hg19 -dbtype snp138 /nfs/srv/databases/ngs/lizaminina/snp.avinput humandb/ | Информация о том, какие SNP имеют rs |
| annotate_variation.pl -out /nfs/srv/databases/ngs/lizaminina/ref -build hg19 /nfs/srv/databases/ngs/lizaminina/snp.avinput humandb/ | Аннотация SNP в базе данных refgene |
| annotate_variation.pl -filter -out /nfs/srv/databases/ngs/lizaminina/dbs -build hg19 -dbtype snp138 /nfs/srv/databases/ngs/lizaminina/snp.avinput humandb/ | Аннотация SNP в базе dbsnp |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out /nfs/srv/databases/ngs/lizaminina/1000g /nfs/srv/databases/ngs/lizaminina/snp.avinput humandb/ | Аннотация SNP в базе данных 1000 genomes. |
| annotate_variation.pl -regionanno -build hg19 -out /nfs/srv/databases/ngs/lizaminina/gw -dbtype gwasCatalog /nfs/srv/databases/ngs/lizaminina/snp.avinput humandb/ | Аннотация SNP в базе данных GWAS |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 /nfs/srv/databases/ngs/lizaminina/snp.avinput -out /nfs/srv/databases/ngs/lizaminina/clin humandb/ | Аннотация SNP в базе Clinvar |