
Последовательность, прочтённую в практикуме 6, можно скачать тут.
Чтобы определить, к какому таксону относится прочтённая последовательность и какую функцию она выполняет, я запустила выравнивание в blastn, используя алгоритм Somewhat similar sequences и прочие настройки - по умолчанию. Поиск проводился по базе данных Nucleotide NCBI. Вот что нашёл blastn:
Прямоугольником выделены лучшие находки (по e-value, весу и покрытию). Flabellina и Moridilla brockii - голожаберные брюхоногие моллюски, которые относятся к общей кладе Aeolididina, поэтому таксономическое положение находки - брюхоногие моллюски клады Aeolididina.

Голожаберный брюхоногий моллюск рода Flabellina
Как можно убедиться по названиям последовательностей в выдаче blastn, прочитанная последовательность кодирует субъединицу 1 митохондриального белка цитохром c-оксидазы, который входит в состав электроно-транспортной цепи митохондрий.
Ниже представлено таксономическое положение родов Flabellina и Moridilla. Выбирался самый младший общий таксон.

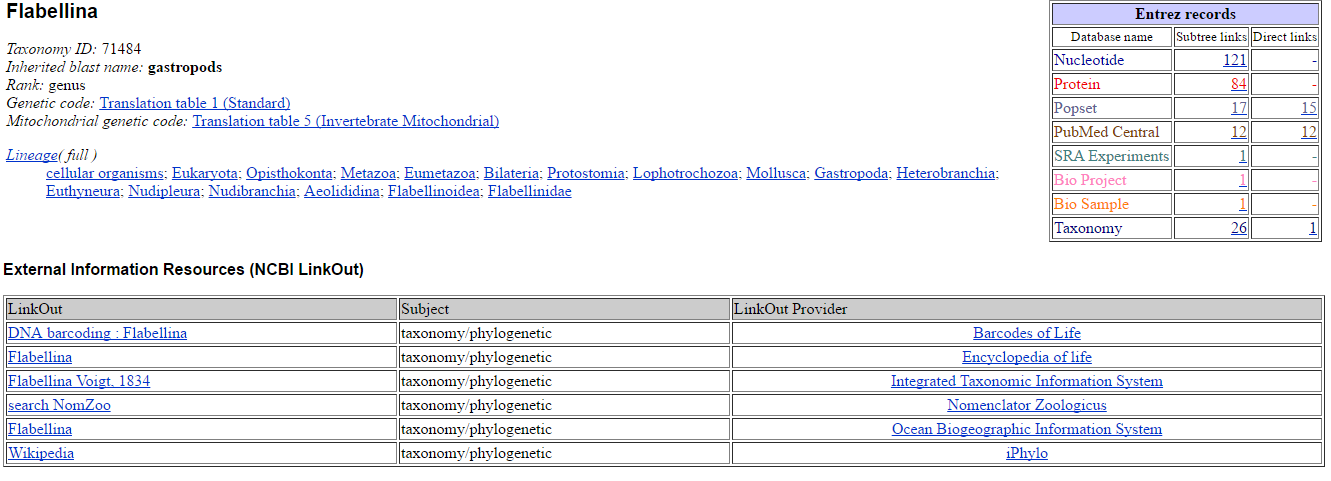
Поиск проводился по роду Flabellina, так как именно к нему относится большая часть находок. Использовалась база данных Nucleotide, все остальные настройки были взяты по умолчанию. Результаты поиска представлены в таблице ниже.
| Алгоритм | Количество находок | E-value лучшей находки | E-value худшей находки | Процент сходства для худшей находки |
| Somewhat similar sequences (blastn) | 65 | 0,0 | 1,4 4e-128 | 100 % (очень короткий ген рРНК) 81 % |
| Discontiguous blast | 64 | 0,0 | 1e-139 | 81 % |
| Megablast | 36 | 3e-165 | 7e-127 | 82% |
Самый точный из представленных алгоритмов - megablast. Он выдал находок меньше, чем два других алгоритма, но эти находки были с лучшими параметрами. Discontiguous megablast выдал большее число находок, однако их можно считать достоверными, так как они относятся к тому же гену, что и заданная последовательность. Blastn дополнительно нашёл последовательность гена рРНК, очень короткую (покрытие 2 %) и с худшим E-value, то есть этот алгоритм может выдавать очевидно недостоверные находки. Таким образом, blastn может выдавать последовательности любой длины, покрытие для него не имеет значения; discontiguous megablast ищет сходные последовательности, потенциально являющимися гомологами, а megablast подходит для поиска очень близких гомологов, так как использует не только E-value, но и процент сходства.

Конец выдачи blastn. Последняя, очевидно недостоверная находка выделена прямоугольником.
Для выполнения этого задания я выбрала следующие три белка, которые, по идее, должны быть у всех эукариот:
| Название (идентификатор UniProt) | Краткое описание |
| H4_HUMAN | Один из пяти эукариотических гистонов - белков, связывающих в клетке двойную спираль ДНК. Гистоны имеются почти у всех эукариот (кроме динофлагеллят) и высококонсервативны. |
| CISY_HUMAN | Цитратсинтаза - один из ферментов цикла Кребса, который есть у большинства аэробных эукариот (в том числе должен быть и у Amoeboaphelidium). |
| PABP2_HUMAN | Белок, связывающий 3'-концевой поли(А)-хвост эукариотической мРНК. У эукариот процессинг мРНК обязательно включает этап добавления поли(А)-хвоста, который регулирует стабильность транскрипта. |
Последовательности всех белков я для удобства собрала в один fasta-файл. Сначала я создала банк данных из последовательности генома организма Amoeboaphelidium. Для этого была использована команда makeblastdb -in X5.fasta -dbtype nucl. Далее требовалось проверить наличие гомологов в геноме данного организма. Задачу можно сформулировать так: требуется провести локальный blast аминокислотной последовательности каждого из белков против трансляции нуклеотидного банка данных (последовательности генома Amoboaphelidium). Этой задаче соответствует алгоритм tblastn, и он запускается командой: tblastn -query proteins.fasta -db X5.fasta -out proteins.out -outfmt 7.
Для каждого белка нашлось ненулевое (что важно!) количество находок разного качества (хорошая - значит с e-value < 0,001). Итоги работы находятся в файле и представлены в таблице ниже:
| UniProt идентификатор | Количество хороших находок/ Общее число находок | Лучшая находка | E-value лучшей находки | Процент идентичности лучшей находки |
| H4_HUMAN | 7/9 | unplaced-368 | 1e-48 | 93,9 |
| CISY_HUMAN | 6/6 | scaffold-693 | 2e-180 | 69,5 |
| PABP2_HUMAN | 11/28 | scaffold-100 | 2e-28 | 62,79 |
При выполнении этого задания я прежде всего получила данные о длинах контигов с помощью команды infoseq X5.fasta -only -name -length. Я выбрала контиг с именем unplaced-665 (его длина была около 20 тысяч п. о., то есть ген вполне может там уместиться). Далее с помощью команды seqret X5.fasta:unplaced-665 -out u665.fasta
я получила последовательность этого контига (файл). После этого я запустила blastn с настойками по умолчанию для этой последовательности. Вот что у меня получилось (показаны лучшие находки):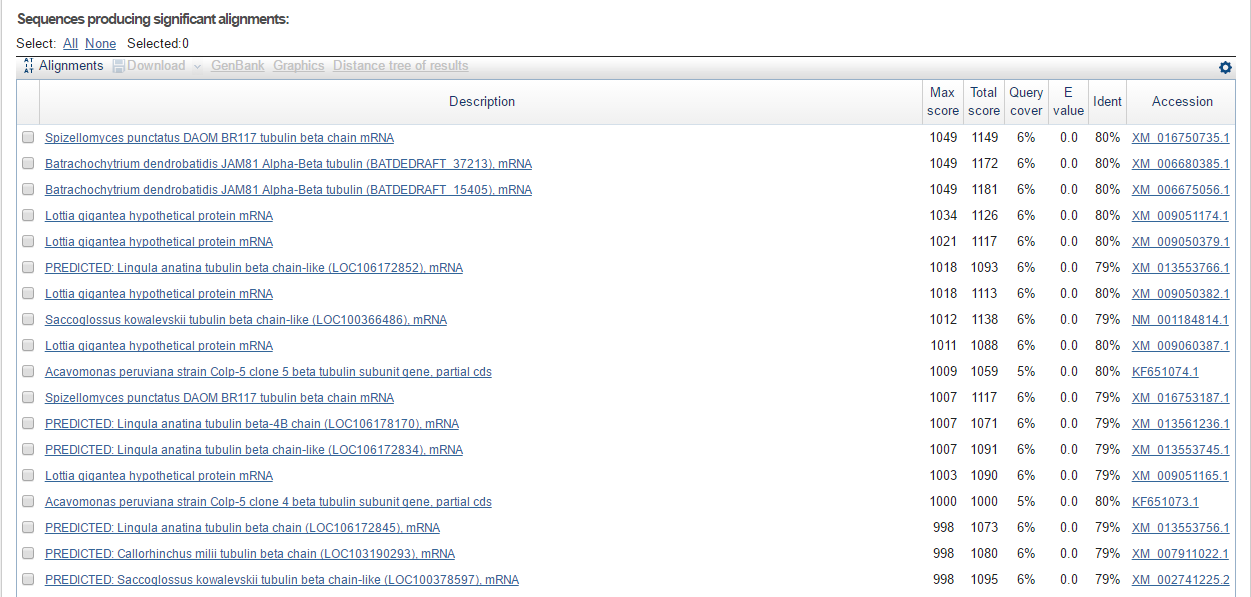
Как можно видеть, все лучшие находки, для которых показано, какой белок они кодируют, кодируют составные части белка тубулина. Покрытие для лучших находок всего 6 %, так что исследуемый контиг содержит ген субъединицы тубулина.