Ниже представлены результаты выполнения упражнений по командам пакета EMBOSS.
Собирает несколько файлов в формате fasta в единый файл в формате fasta. Я собрала в один файл все белки, у которых АС в Uniprot начинается с E11. Результат.
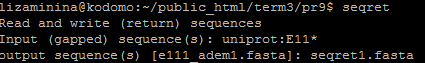
В seqret можно подать несколько последовательностей и другим способом. Я создала исходный файл, содержащий имена последовательностей USA и подала его в команду seqret. В итоге все три последовательности были переписаны в формате fasta в итоговом файле. Команда представлена на рис.:
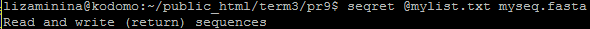
Команда seqretsplit один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы. Исходный файл. Результат: файл 1, файл 2, файл 3.
Из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле. Файл с хромосомой, результат. Был создан рабочий файл, который и подавался на вход программе.

Команда transeq транслирует кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле. Исходный файл, результат.
Транслировать данную нуклеотидную последовательность в шести рамках. Исходный файл, результат.
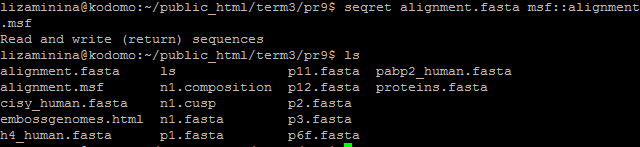
Перевести выравнивание из fasta формате в формат .msf. Исходный файл, результат.
infoalign выводит число совпадающих букв между второй последовательностью выравнивания (опция -refseq 2) и всеми остальными (на выходе только имя последовательности и число: опции -only -name -idcount). Исходный файл, результат.

Перевести аннотации особенностей в записи формата .gb в табличный формат .gff. Исходный файл, результат.

Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями. Исходный файл, результат
Команда shuffle перемешивает буквы в данной нуклеотидной последовательности. Исходный файл, результат см. рис.
Команда cusp находит частоты кодонов в данных кодирующих последовательностях. Исходный файл, результат.
Команда compseq находит частоты динуклеотидов в данной нуклеотидной последовательности и сравнивает их с ожидаемыми. Исходный файл, результат.

Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов. Я использовала последовательности гистона Н4 (файл) и их выравнивание, построенное с помощью алгоритма Muscle Jalview. Результатом было выравнивание нуклеотидных последовательностей.
Для выполнения этого задания я выбрала геномы двух видов бактерий, относящихся к одному роду: Corynebacterium diphtheriae и Corynebacterium efficiens. Для построения выравнивания я использовала инструмент blast2seq blastn, который строит выравнивание двух заданных последовательностей. Использовались параметры алгоритма по умолчанию. Одним из результатов работы программы является карта локального сходства, представленная на рисунке ниже.
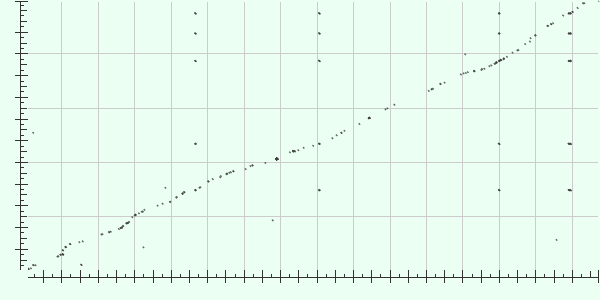
По горизонтали отложен геном Corynebacterium efficiens, по вертикали геном Corynebacterium diphtheriae. Как можно видеть, в целом график очень близок к прямой линии, что свидетельствует о близости двух видов. Изредка выпадают некоторые нуклеотиды, что позволяет судить о том, что эти виды, несмотря на эволюционную близость, всё-таки отличаются друг от друга.
Более интересная картина наблюдается при выравнивании хромосом II бактерий Brucella melitensis biovar Abortus (ID NC_007624.1) и Brucella suis (ID NC_004311.2).
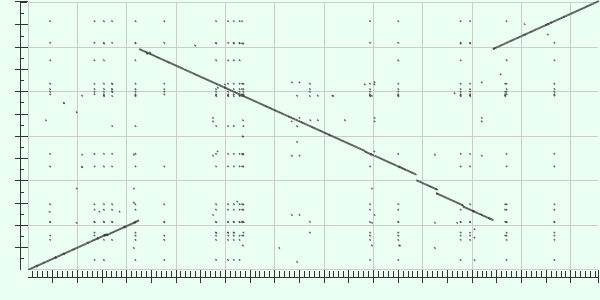
По горизонтали отложен геном Brucella melitensis, по вертикали Brucella suis. Можно видеть довольно крупный участок, идущий перпендикулярно основной линии. По-видимому, он соответствует крупной инверсии. Рядом с ним можно увидеть ещё два небольших отрезка, перпендикулярных основной линии, которые, вероятно, отражают более мелкие инверсии.