1. Моделирование эволюции белка YPWA и выравнивание c мутантами
(нереалистичные параметры)
Скрипт
evolve_protein.pl создает мутантные копии белка; вариабельными элементами скрипта
являются вероятность ошибки ДНК-полимеразы и вероятность (в случае ее ошибки) инсерции или делеции.
| -i |
Имя открываемого файла |
| -o |
Имя создаваемого файла |
| -c |
Вероятность ошибки ДНК-полимеразы |
| -r |
Вероятность замены аминокислоты на другую аминокислоту в случае ошибки ДНКП |
| -g |
Количество поколений |
| -f |
Заставляет скрипт создавать полноразмерные последовательности; |
| -t |
Задает количество последовательностей, которые нужно создать |
В результате работы скрипта были созданы 3 файла. Они содержали последовательности с разными параметрами -c и -r трех мутантов; из последовательностей случайным образом вырезались 20-аминокислотые пептиды.
После создания файлов в программе JalView было найдено оптимальное выравнивание для каждого мутанта. В таблице 1 приведены
данные по различным параметрам сравнения двух последовательностей в выравнивании.
| Табл.1. Сравнение попарных выравниваний. Одним цветов раскрашены аминокислоты, близкие по свойствам. |
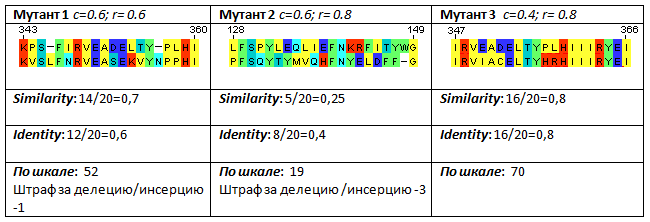 |
Самым большим
весом по шкале замен с исходным белком обладает мутант №3, это обусловлено самой низкой
вероятностью ошибки ДНК-полимеразы (0,4) и, в случае ее ошибки, низкой вероятностью исчезновения или
появления нового аминокислотного остатка. Кроме того, заметно, что вероятность появления вставки/делеции
различается; у первого мутанта – с самой большей вероятностью возникновения гэпа или инсерции – в процессе
«эволюции» появилось 2 вставки, тогда как у двух других мутантов их число меньше (аналогичная вероятность всего
0,2 при условии ошибки ДНКП). Поэтому было принято решение штрафовать за инсерции или делеции в первом случае менее строго,
нежели за появление оных в мутантов 2 и 3.
Similarity: мутант 2 имеет самый низкий уровень похожести, это достигалось за счет высокой
вероятности ошибки ДНКП и замены аминокислот. Так как вероятность вставки/гепа не самая высокая, то и штраф за
него должен быть больше, чем штраф а них у первого мутанта. Самый высокий уровень сходства с исходным белком имеет
мутант 3, это согласуется с тем, что у него довольно низкий уровень вероятности ошибки ДНКП.
Identity: по вышеизложенным причинам мутант 3 имеет самый высокий уровень идентичности с исходным
белком; разница между двумя другими мутантами объясняется тем, что во втором случае понижена вероятность замены
исходного а.о. при ошибке ДНКП на такой же за счет того, что по сравнению с первым мутантом увеличилась вероятность
вставки или делеции.
2. Выравнивание YPWA_BACSU с ортологами
Был создан
файл в fasta-формате с тремя последовательностями, включая последовательность YPWA_BACSU.
С помощью программы Muscle произведено выравнивание последовательностей, а также раскрашивание по схеме ClustalX. Пороговый процент идентичности - 66% (для выделения
аминокислот, совпадающих как минимум в двух из трех последовательностей. Файл сохранен
здесь.
О каждой паре последовательностей была получена информация с помощью команды
infoalign. Как и для других команд пакета EMBOSS,
для нее существуют некоторые параметры.
INPUT и OUTPUT команда спрашивает самостоятельно.
| -matrix |
Матрица весов замен, которая будет использоваться при сравнении последовательностей. По
умолчанию для белковых последовательностей используется матрица EBLOSUM62, для нуклеотидных
последовательностей матрица EDNAFULL |
| -html |
Вывод данных в виде кода html-таблицы |
| -refseq |
Последовательность, номер или название которой было введено, становится последовательностью, относительно которой сравниваются другие последовательности.
По умолчанию такой последовательностью является консенсусная |
| -help |
Помощь по команде |
| -only |
Позволяет ограничивать тип выводимой информации, требует дополнительных указаний на требуемые параметры |
Следующие флаги работают только с
-only, выводя следующие типы данных:
| -heading |
Показывать заголовки колонок |
| -usa |
USA (Uniform Sequence Address) |
| -seqlength |
Длина сравниваемых последовательностей |
| -alignlength |
Длина выровненных последовательностей (с гэпами) |
| -gaps |
Количество пропусков |
| -gap |
Количество пропусков |
| -idcount |
Количество идентичных позиций |
| -simcount |
Количество позиций со схожими по свойствам аминокислотами |
| -diffcount |
Количество позиций с разными аминокислотами |
| -change |
Количество измененных позиций |
| -weight |
Показывать колонку с весами выравнивания |
| -description |
Описание |
Полученные данные можно посмотреть в таблицах 2, 3 и 4.
| Табл. 2. Информация о выравнивании 1 |
| Name |
Sequence Length |
Aligned Length |
Gaps |
Gap Length |
Identity |
Similarity |
Difference |
% Change |
Weight |
Description |
| E0RBL5_PAEP6/1-505 |
505 |
505 |
0 |
0 |
505 |
0 |
0 |
0.000000 |
1.000000 |
Hypothetical metalloprotease OS=Paenibacillus
polymyxa (strain E681) GN=PPE_02619 PE=4 SV=1 |
| P50848/1-501 |
501 |
505 |
2 |
4 |
249 |
102 |
150 |
50.693069 |
1.000000 |
metal-dependent carboxypeptidase |
|
| Табл. 3. Информация о выравнивании 2 |
| Name |
Sequence Length |
Aligned Length |
Gaps |
Gap Length |
Identity |
Similarity |
Difference |
% Change |
Weight |
Description |
| E0RBL5_PAEP6/1-505 |
505 |
505 |
0 |
0 |
505 |
0 |
0 |
0.000000 |
1.000000 |
Hypothetical metalloprotease OS=Paenibacillus polymyxa (strain E681) GN=PPE_02619 PE=4 SV=1 |
| C3C074_BACTU/1-505 |
505 |
505 |
0 |
0 |
261 |
89 |
155 |
48.316833 |
1.000000 |
Metalloprotease OS=Bacillus thuringiensis serovar tochigiensis BGSC 4Y1 PE=4 SV=1 |
|
| Табл. 4. Информация о выравнивании 3 |
| Name |
Sequence Length |
Aligned Length |
Gaps |
Gap Length |
Identity |
Similarity |
Difference |
% Change |
Weight |
Description |
| P50848/1-501 |
501 |
505 |
2 |
4 |
501 |
0 |
0 |
0.792079 |
1.000000 |
metal-dependent carboxypeptidase |
| C3C074_BACTU/1-505 |
505 |
505 |
0 |
0 |
311 |
78 |
116 |
38.415840 |
1.000000 |
Metalloprotease OS=Bacillus thuringiensis serovar tochigiensis BGSC 4Y1 PE=4 SV=1 |
|
3. Модель эволюции белка YPWA и выравнивание c мутантами
(ситуация с реалистичными параметрами)
Задание, аналогичное первому, но с другими параметрами, было проделано для белка YPWA снова. Были получены данные,
представленные в таблице 5.
| Табл.5. Сравнение попарных выравниваний. Одним цветов раскрашены аминокислоты, близкие по свойствам. |
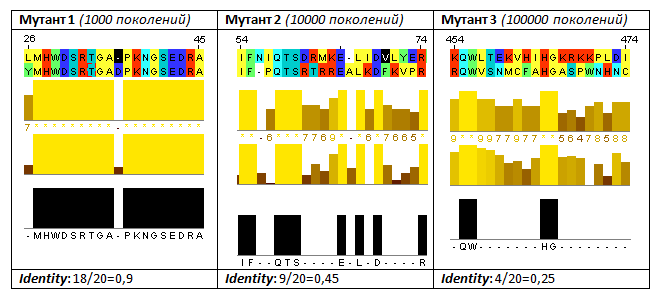 |
При анализе этой таблицы были сделаны следующие выводы.
Если условия использования скрипта максимально приближены к реальности, наблюдаются
следующие тенденции: гораздо рее встречаются замены аминокислот, эволюция белка происходит не очень быстро.
Однако если учесть скорость размножения микроорганизмов, темпы накопления изменений в белке становятся
заметны даже на относительно небольшом промежутке времени. Так, 1000 поколений проходит за 30 000 минут,
то есть за 500 часов, то есть всего за 20,8 суток; соответственно, 10 000 поколений пройдут за 208 суток,
то есть приблизительно за 7 месяцев. А промежуток, необходимы для кардинального изменений последовательности
– только 5,7 лет. За это время некоторые организмы даже половозрелыми не становятся.