Сборка генома de novo
Скачивание чтений
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR424/001/SRR4240361/SRR4240361.fastq.gz
Краткая информация про чтения:
Организм: Buchnera aphidicola str. Tuc7 (Acyrthosiphon pisum)
AC: SRR4240361
Прибор: Illumina Genome Analyzer II
Тип чтений: одноконцевые (SE)
Подготовка чтений
Удаление адаптеров
Я скопировала все адаптеры и объединила их в один файл: cat *fa > adapters.fasta
Теперь запускаю trimmomatic для SE с функцией ILLUMINACLIP:adapters.fasta:2:7:7
java -jar /usr/share/java/trimmomatic.jar SE -threads 10 SRR4240361.fastq.gz SRR4240361_noadap.fastq.gz ILLUMINACLIP:adapters.fasta:2:7:7 2> adap_logs.txt
Загляну внутрь файла с логами:
На вход дано 7272621 чтений, удалено 34532 (0.47%). То есть 0.47% чтений оказались остатками адаптеров.
Обрезание чтений
java -jar /usr/share/java/trimmomatic.jar SE -threads 10 SRR4240361_noadap.fastq.gz SRR4240361_trim.fastq.gz TRAILING:20 MINLEN:32 2> trim_logs.txt
Загляну внутрь файла с логами:
На вход дано 7238089 чтений, удалено 403754 (5.58%), осталось 6834335 чтений.
Размер файлов:
SRR4240361.fastq.gz - сырые чтения - 193M
SRR4240361_noadap.fastq.gz - чтения без адаптеров - 192M
SRR4240361_trim.fastq.gz - триммированные чтения - 178M
Подготовка k-меров
velveth velveth/ 31 -fastq.gz -short SRR4240361_trim.fastq.gz
velveth/ - папка, в которую будут загружаться файлы на выходе программы
31 - длина k-меров
-fastq.gz - указываем, что файл на вход в формате .fastq.gz
-short - короткие и непарные чтения
SRR4240361_trim.fastq.gz - файл с чтениями (триммированными)
Сборка k-меров
velvetg velveth/
velveth/ - папка с результатами работы velveth
Из вывода программы в stdout получаем следующую информацию
N50 = 25683
max 49238
Посмотрим файл stat.txt. Он состоит из столбцов, разделенных tab, во втором столбце - length, поэтому 3 самые длинные контига можно с помощью:
cut -f2 stats.txt | sort -h | tail -3
И по ним найдем целые строки с этими контигами. Получим:
| ID | length | coverage |
|---|---|---|
| 6 | 49238 | 26,66 |
| 2 | 45555 | 26,45 |
| 34 | 43866 | 23,51 |
Чтобы посмотреть на аномальное покрытие, пишу команду:
cut -f6 stats.txt | sort -h
Аномально большими покрытиями являются, например, значения 561.0, 865.0 и 212829.0; аномально маленькими - от 1 до 5. При этом заметно, что контиги с аномальными покрытиями (и большими, и маленькими) имеют длину меньше 31 (то есть заданного k-мера). Насколько я понимаю, такие контиги не попадают в contigs.fa, но их можно посчитать, например:
Посчитать, сколько попало контигов в contigs.fa, можно так:
grep '^>' contigs.fa | wc -l
Ответ: 208 контигов (длиной 31 и больше)
Анализ
Чтобы получить fasta-файлы трех самых крупных контигов, я разделила contigs.fa на отдельные fasta-файлы:
seqretsplit -filter contigs.fa dir/name.format
Теперь буду использовать три самые крупные контига и сравню их с хромосомой Buchnera aphidicola (GenBank/EMBL AC — CP009253) в megablast
Самый длинный контиг
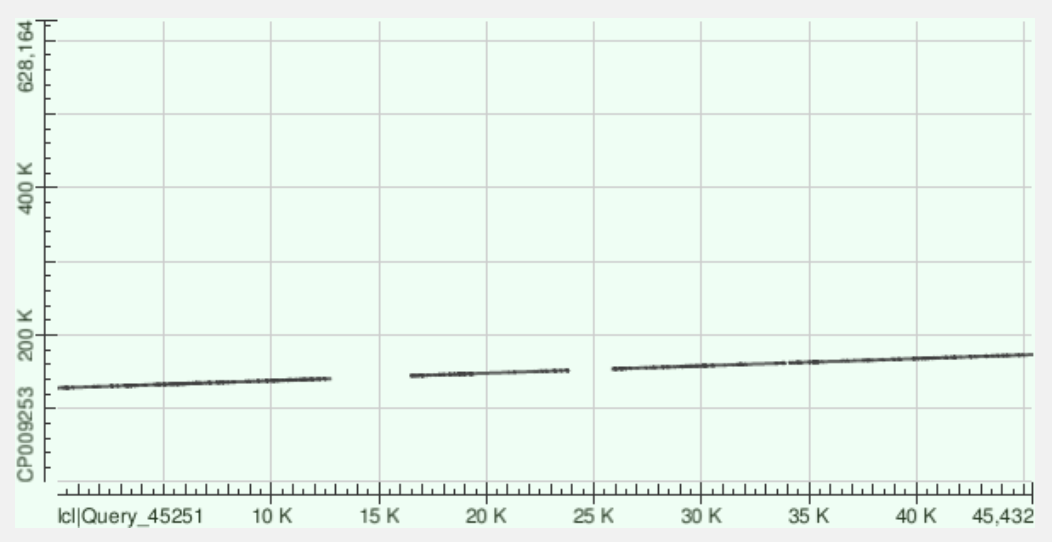
По DotPlot видно, что между выровненными участками происходили делеции и мутации.
| коорд контига | коорд хромосомы | SNP | гэпы | Identity |
|---|---|---|---|---|
| 50-12790 | 127825-140555 | 3259 | 548 | 75% |
| 16429-23828 | 144368-151796 | 1677 | 243 | 78% |
| 25809-33893 | 153752-161738 | 1813 | 264 | 78% |
Второй самый длинный контиг
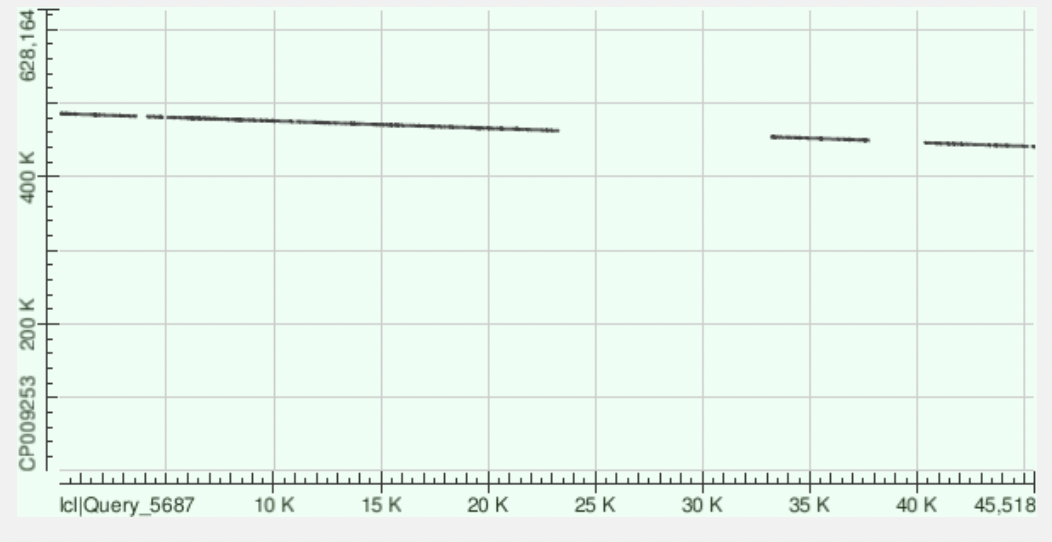
По DotPlot видно, что наклон отрицательный, то есть запись контига перевернута. Кроме того, видно, что между первыми двумя крупными совпадающими участками произошла крупная делеция, а между вторым и третим крупными участками находится негомологичный участок.
Вообще, megablast выделил 9 гомологичных участков. Укажу только координаты
на контиге и %Identity:
440755:440944 - 89%
441135:442817 - 79%
442877:445895 - 80%
449411:454069 - 75%
462496:467421 - 77%
467412:474667 - 77%
474844:480660 - 74%
480874:481545 - 82%
481997:485679 - 77%
Процент гэпов варьируется от 2 до 4.
Третий самый длинный контиг
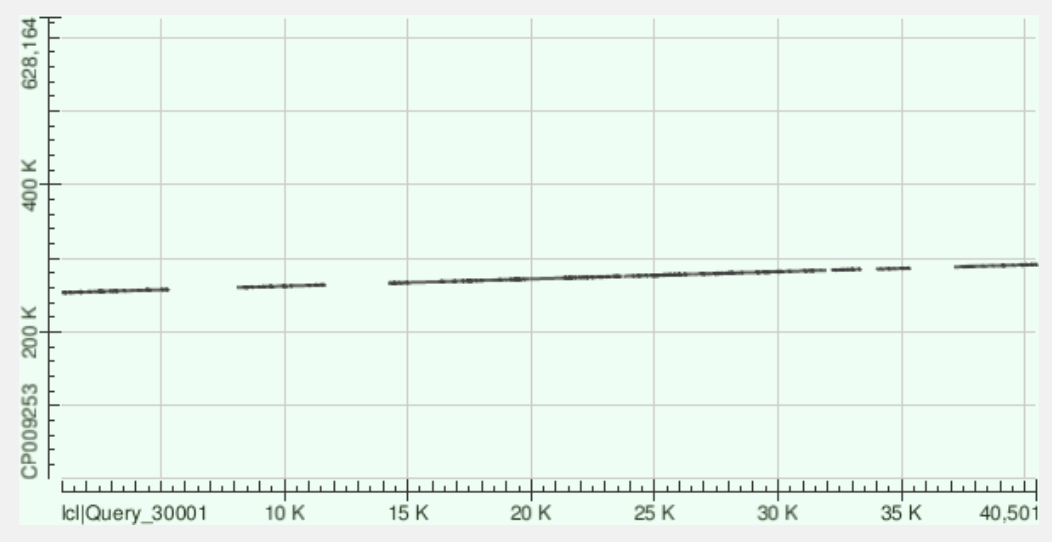
По Dotplot видно, что между гомологичными участками произошли делеции.
Вообще, megablast выделил 7 гомологичных участков. Укажу только координаты
на контиге и %Identity:
253223:257546 - 73%
260224:263784 - 77%
266073:275551 - 79%
275566:283706 - 76%
283963:285070 - 76%
285200:286535 - 76%
288181:291560 - 78%
Процент гэпов варьируется от 2 до 5.