Практикум 12
Множественное выравнивание последовательностей белков
Сравнение выравнивания одних и тех же последовательностей разными программами
Я взяла белки из практикума 10
OS: Bunyamwera virus (BUNV)
ID: GP_BUNYW
AC: P04505
Выровняла их с помощью разных программ. Ниже представлено сравнение выравниваний
Сравнение Clustal и Muscle
Доля одинаково выровненных позиций в первом выравнивании: 0.84%
Доля одинаково выровненных позиций во втором выравнивании: 0.83%
По выдаче программы можно понять, что до 27 позиции выравнивания не совпадают. Это можно увидеть на представленной ниже картинке, которая была сделана на сайте VerAlign

Сравнение Clustal и Mafft
Доля одинаково выровненных позиций в первом выравнивании: 0.82%
Доля одинаково выровненных позиций во втором выравнивании: 0.8%
Данные выравнивания также не совпадают до 27 позиции

Сравнение TCoffee и Mafft
Доля одинаково выровненных позиций в первом выравнивании: 0.85%
Доля одинаково выровненных позиций во втором выравнивании: 0.84%
Тут мы уже можем заметить, что они не совпадают до 23 позиции (она совпадает с 19 во втором выравнивании)

Сравнение TCoffee и Muscle
Доля одинаково выровненных позиций в первом выравнивании: 0.86%
Доля одинаково выровненных позиций во втором выравнивании: 0.86%
Тут выравнивания не совпадают с 848 по 863 позицию

Сравнение TCoffee и Clustal
Доля одинаково выровненных позиций в первом выравнивании: 0.83%
Доля одинаково выровненных позиций во втором выравнивании: 0.84%
Выдает такой же результат, так и при сравнении Clustal и Muscle, потому что выравнивания Muscle and TCoffee практически идентичны, но совпадения начинаются только с 32 позиции

Сравнение Muscle и Mafft
Доля одинаково выровненных позиций в первом выравнивании: 0.91%
Доля одинаково выровненных позиций во втором выравнивании: 0.9%
Эти программы имеют наибольшее сходство. Совпадения начинаются уже с 9 позиции, однако выпадает блок 523-546

Ссылки на файлы с выравниванием в FASTA формате:
Построение выравнивания по совмещению структур и сравнение его с выравниванием MSA
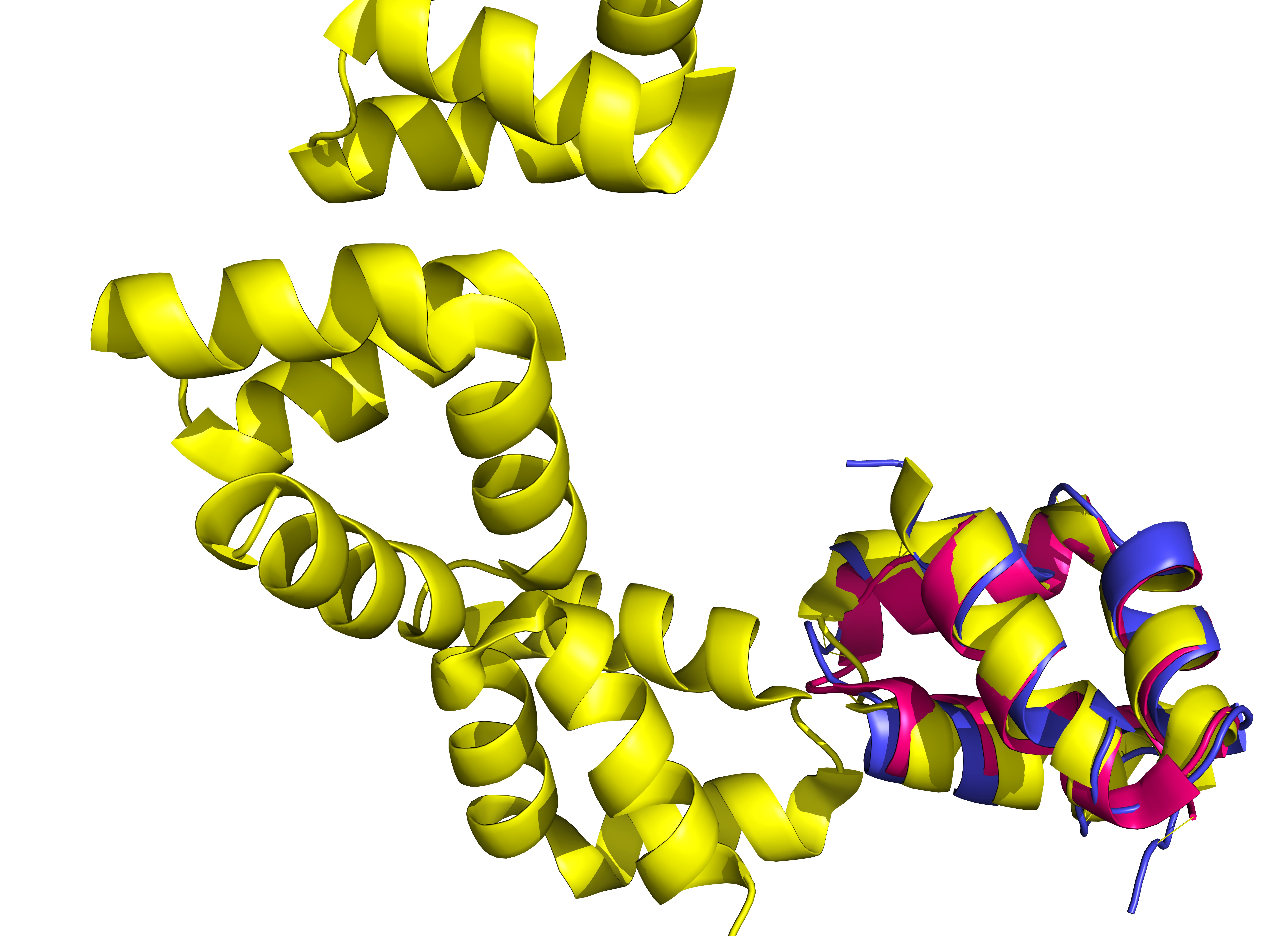
Для выполнения выравнивания я выбрала семейство Bacteriocin_IIi с Pfam AC: PF11758 . Затем нашла 3D структуры белков этого семейства, выбор пал на белки с PDB ID: 2N8P , 2N8O ,6SIG . Было построено выравнивание на сайте PDB, в Jalview и рассмотрены совмещенные 3D структуры белков в PyMol. Если сравнивать выравнивание на сайте PDB и сделанное в Jalview (проект), то можно сказать, что выравнивание с помощью второй программы обладает наибольшей точностью.

Так же в выравнивании, скаченном с сайта PDB (ссылка) мы можем заметить появление «лишних» аминокислот в начале, их можно увидеть на модели, построенной в PyMol (розовый и фиолетовый концы на Риc.8 и Рис.9). Все выравнивания подтверждают высокий уровень сходства псоледовательностей.
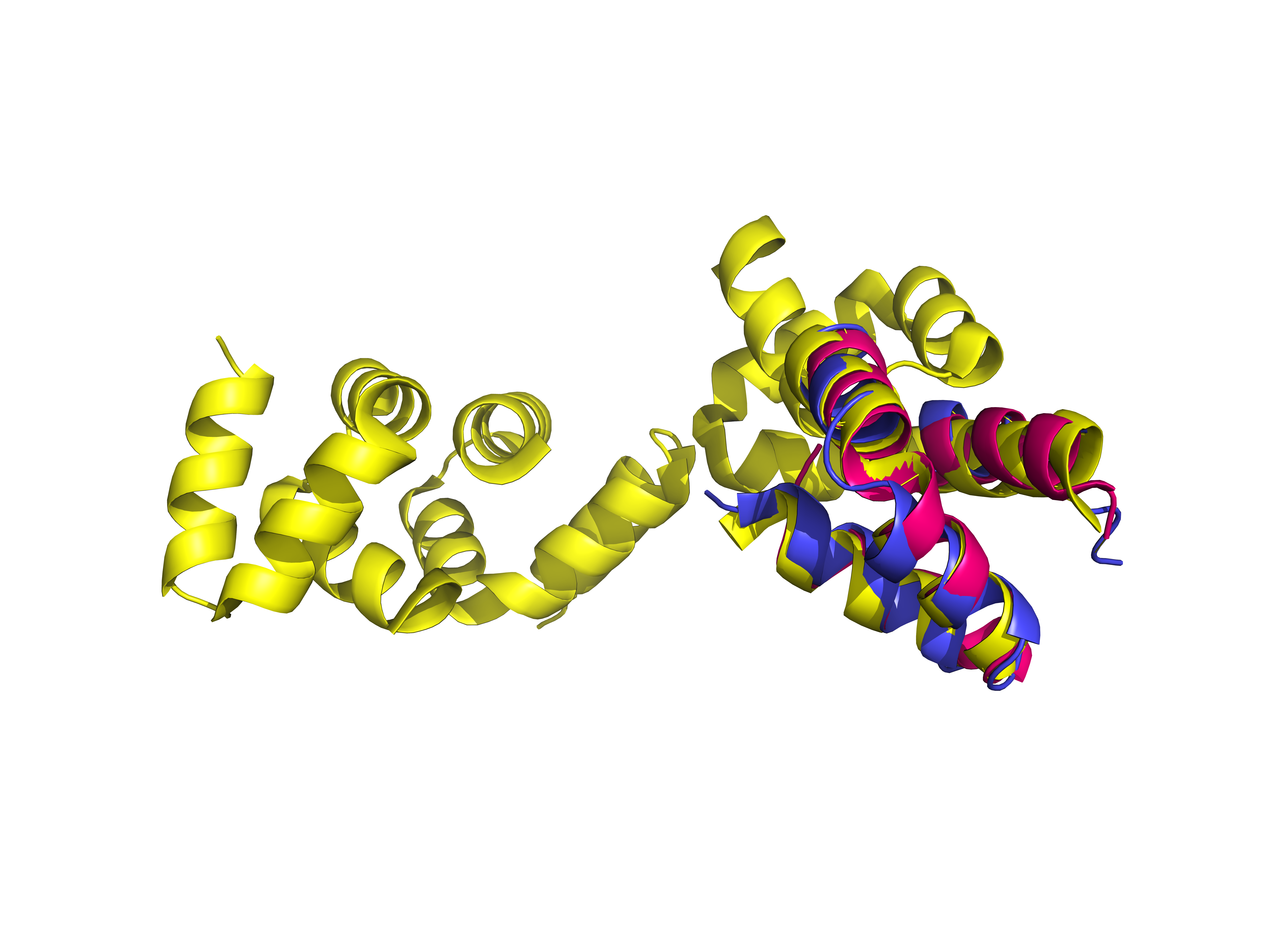
Краткое описание одной из программ MSA
Мно́жественное выра́внивание после́довательностей (multiple sequence alignment, MSA) — выравнивание трёх и более биологических последовательностей, обычно белков, ДНК или РНК. В большинстве случаев предполагается, что входной набор последовательностей имеет эволюционную связь. Используя множественное выравнивание, можно оценить эволюционное происхождение последовательностей, проведя филогенетический анализ.
T-Coffee:
T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation) имеет две основные особенности.Во-первых, она обеспечивает простое и легко выполнимое средство генерации множества выравниваний с использованием разнородных источников данных. Данные из этих источников предоставляются T-Coffee через библиотеку парных выравниваний.Второй основной особенностью T-Coffee является метод оптимизации, который используется для поиска множественного выравнивания, которое наилучшим образом соответствует парным выравниваниям во входной библиотеке с использованием прогрессивной стратегии, которую можно сравнить с той, которая используется в ClustalW. Преимущество метода оптимизации заключается в том, что он быстрый и надежный. Информация в библиотеке используется для выполнения прогрессивных выравниваний и облегчает обязанность учитывать выравнивания между всеми парами при выполнении каждого шага прогрессивных выравниваний.
Источники: