Исследование структуры тРНК
- Краткое описание структуры в файле 1U0B.pdb В файле приведены координаты атомов следующих молекул
- Цистеинил-тРНК-синтетаза;
- Цистеинил-тРНК
Структура выделена из Еrichia coli.
Для исследования была выбрана цепь A, представляющая Цистеинил-тРНК со следующей последовательностью:
[1] 5' -GGCGCGUUAACAAAGCGGUUAUGUAGCGGAUUGCAAAUCCGUCUAGUCCGGUUCGACUCCGGAACGCGCCUCCA-3' [76],
где 1 и 76 - номера первого и последнего нуклеотида (нуклиотиды 17 и 47 в файле pdb не указаны).B последовательности на 3'-конце есть триплет CCA, к которому присоединяется аминокислота, приведены координаты его атомов.
- Исследование вторичной структуры
С помощью программы find_pair пакета 3DNA были определены возможные водородные связи
между азотистыми основаниями. Увидеть выходной файл программы можно здесь. В соответствии с полученными данными:
- акцепторный стебель состоит из участка 1-7 и комплементарного ему участка 66-72;
- Т-стебель состоит из участка 49-53 и комплементарного ему участка 61-65;
- D-стебель состоит из участка 10-12 и комплементарного ему участка 23-25;
- антикодоновый стебель состоит из участка 38-44 и комплементарного ему участка 26-32.
Структуру стеблевых дуплексов поддерживают 25 канонических и 6 неканонических пар оснований.Рис.1. Вторичная структура Цистеинил-тРНК из Еrichia coli 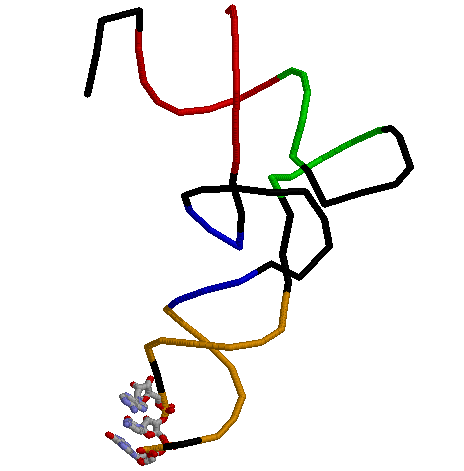
Скрипт для получения изображения
background white restrict RNA wireframe off color black backbone 100 select (1-7, 66-72) and RNA color red select (49-53, 61-65) and RNA color green select (10-12, 23-25) and RNA color blue select (38-44, 26-32) and RNA color orange select (34, 35, 36) and RNA backbone off wireframe 100 cpk 150 color cpk
Даный скрипт служит для получения в RasMol изображения остова исследуемой тРНК, где акцепторный стебель выделен красным, Т-стебель - зеленым, D-стебель - синим, антикодоновый - оранжевым.
В шарнирной модели представлен антикодон (GCA)к цистеину.
Неканоническая пара - А-А (46-й и 14-й нуклеотиды)
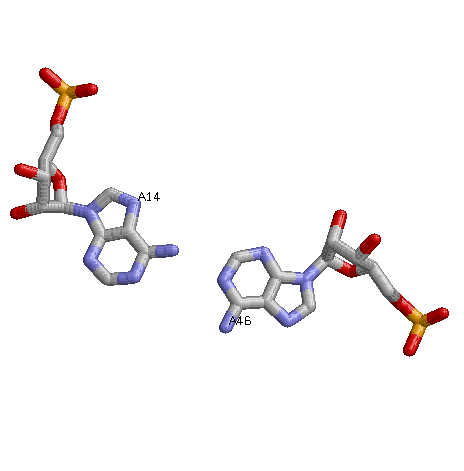
- вариабельная петля отсутствует
- тимидин в Т-петле отсутствует
- дигидроуридинов в D-петле нет;
- Исследование третичной структуры
1. Программа analiyze показала предполагаемые стекинг-взаимодействия между парами оснований, связанных водородными связями. Всего 30 возможных стекинг-взаимодействий (31 пара оснований) Из них были выбраны взаимодействия номер 7 и 22.
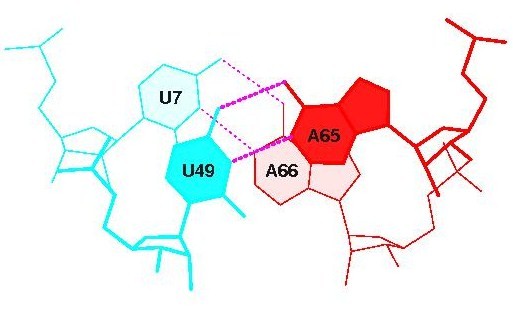
Стекинг-взаимодействия между основаниями конца акцепторно стебля и начала Т-стебля:
Стекинг-взаимодействия между антикодоновым и D-стеблем:
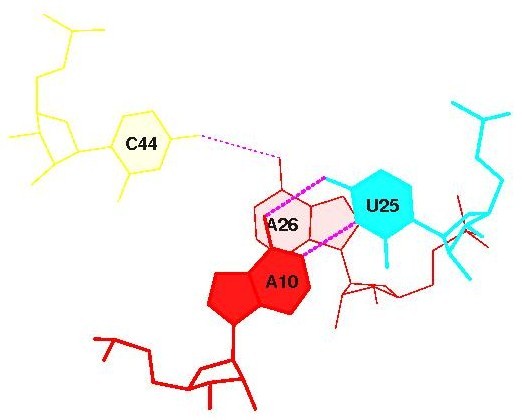
суммарная площадь перекрывания в первом случае равна 1.54-м квадратным ангстреммам, в другом 1.36-ти. Это не очень много по сравнению с другими взаимодействиями (максимально 14), но всё же, это единственные взаимодействия на участках, которые требовалось рассмотреть в задании. 2.
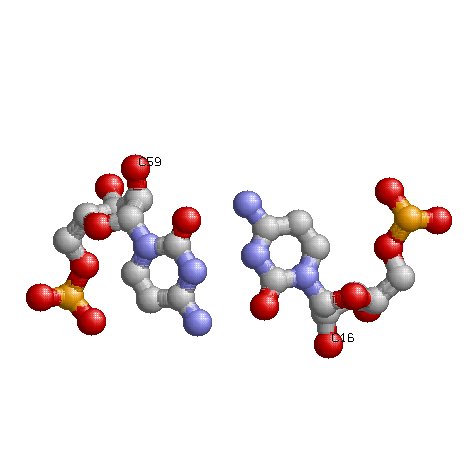
Дополнительные водородные связи между основаниями D- и Т-петель есть. Они образованы нуклеотидами:
D-петля Т-петля G15 G48 C16 C59 G19 C56 Первые 2 пары неканонические, 3-я каноническая.
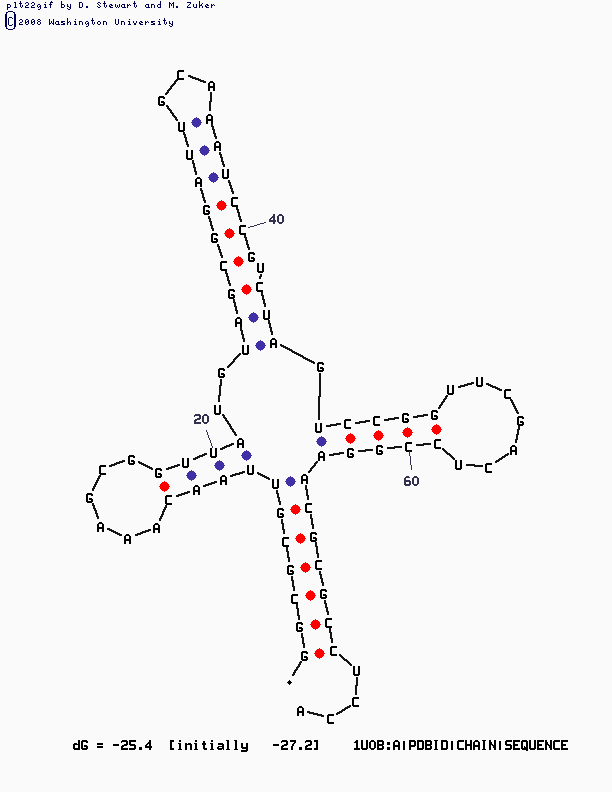
- Предсказание вторичной структуры тРНК
Участок структуры
Позиции в структуре
(по результатам find_pair)Результаты предсказания
с помощью einvertedРезультаты предсказания
по алгоритму ЗукераАкцепторный стебель 5' 1-7 3'
5' 66-72 3'
Всего 7 парпредсказанo 8 пар предсказано 7 пар D-стебель 5' 10-12 3'
5' 23-25 3'
Всего 3 парыне предсказано ни одной пары предсказано 4 пары, но, увы, они не совпадают с тем, что есть на самом деле T-стебель 5' 49-53 3'
5' 61-65 3'
Всего 5 парне предсказано ни одной пары предсказаны все 5 пар Антикодоновый стебель 5' 38 - 44 3'
5' 26-32 3'
Всего 7 парпредсказано правильно 5 пар предсказано 9 пар, из них 5 правильно Общее число канонических пар нуклеотидов 21 12 17 в оптимальной структуре Предсказание EINVERTED
Программа не произвела на меня особого впечатления. Конечно, она автоматически заменяет урацил на тимин в последовательности, но предсказание взаимодействий ведётся в предположении, что цепь может перегибаться только в одном месте, ближе к середине. Значит всегда хорошо отыскивается акцепторный стебель и чуть хуже антикодоновый, но отыскать D- или Т-стебель мне, сколько я ни пыталась, не удалось. Судя по алгоритму программы, и не удастся.
Предсказание MFOLD
Программа mfold работатает намного лучше,все результаты можно посмотреть и, видимо, она более, чем первая, настроена на структуру именно РНК. Если запустить программу со входным параметром Р, равным 0 (параметр показывает, на сколько процентов то, что выдаст программа, может отличаться по энергии от оптимального варианта), то появятся3 структуры. Эти же 3 структуры программа предлагала и при всех остальных параметрах.Чем выше значение параметра, тем больше структур предлагает программа. Я остановилась при Р=20. Ни в одной из структур не была правильно предсказана D-петля, поэтому я посчитала наиболее походящей на оригинал самую первую выданную мне структуру. Необходимо учитывать, что нумерация остатков несколько отличается (в последовательности 74 остатка, тогда как в файле pdb их 76, 17-й, 47-й отсутствуют).
©ХАЧАТРЯН ЛУСИНЕ, 2008