Bизуальный анализ движений молекул
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_pbc_1.pdb -skip 20 -pbc mol На вопрос о выводе групп отвечаем Protein
Для того, чтобы открыть pep_pbc_1.pdb в PyMol, скачаем его с суперкомпьютера:
scp -r pep_pbc_1.pdb lynx@kodomo.fbb.msu.ru:lynx/md
Для анализа сруктуры в PyMol необходимо включить анимацию. Результат визуализации неудовлетворителен: молеклу "болтает" по всему экрану. Поэтому введем:
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_fit_1.pdb -skip 20 -fit rot+trans На вопрос о выводе групп отвечаем Protein
| Номер модели и время | 1, t=0.00000 | 5, t=800.00000 | 10, t=1800.00000 | 15, t=2800.00000 | 20, t=3800.00000 |
| Модель |  |
 |
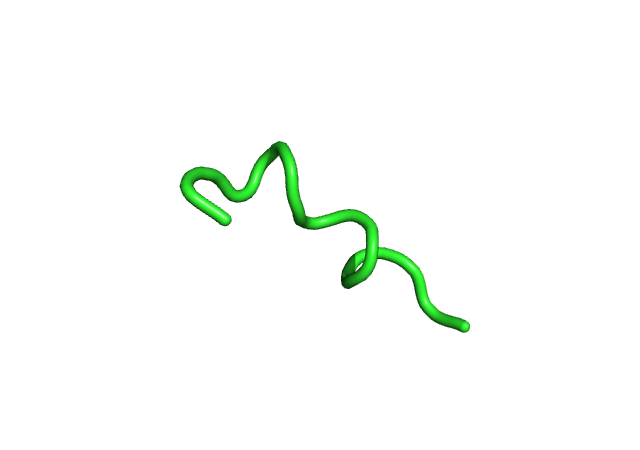 |
 |
 |
| Наблюдения | α-спираль, 4 оборота | Начинает плавиться N-конец | Начинает плавиться С-конец | Наиболее расплавленные структуры, у α-спирали 2-3 оборота | |
| Номер модели и время | 25, t=4800.00000 | 30, t=5800.00000 | 35, t=6800.00000 | 40, t=7800.00000 | 45, t=8800.00000 |
| Модель |  |
 |
 |
 |
 |
| Наблюдения | Наиболее расплавленные структуры, у α-спирали 2-3 оборота | ||||
| Номер модели и время | 50, t=9800.00000 | 55, t=108000.00000 | 60, t=118000.00000 | 65, t=128000.00000 | 70, t=138000.00000 |
| Модель |  |
 |
 |
 |
 |
| Наблюдения | Наиболее расплавленные структуры, у α-спирали 2-3 оборота | ||||
| Номер модели и время | 74, t=14600.00000 | ||||
| Модель |  |
||||
| Наблюдения | α-спираль доконца не восстанавливается | ||||
Для опредления соотвествия между номером модели и временем моделирования надо найти в pdb файле выражение "MODEL 50" (цифра 50 это и есть номер модели) и двумя строчками выше будет упоминание о времени моделирования.
Определение средне-квадратичного отколнения в ходе моделирования
Так как у нас происходит конформационный переход, сначала расчитаем отклонение в ходе все симуляции относительно стартовой структуры:g_rms -f pep_md.xtc -s pep_md.tpr -o rms_1 На вопрос о выводе групп отвечаем Backbone
Значения средне-квадратичного отколнения:
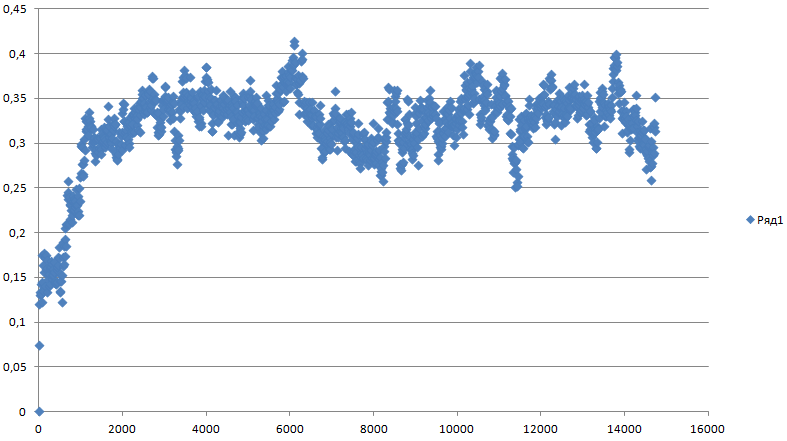
И относительно каждой предыдущей структуры на растоянии 400 кадров (eсли ближе к концу закончился конформационный переход, то отколнение должно уменьшаться):
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_2 -prev 400 На вопрос о выводе групп отвечаем Backbone
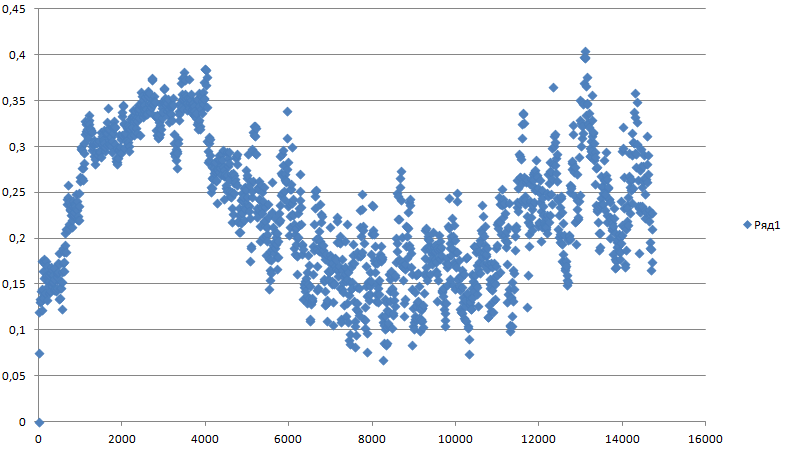
Отклонение действительно уменьшилось.
Определение изменения гидрофобной и гидрофильной поверхности в ходе конформационного перехода
g_sas -f pep_md.xtc -s pep_md.tpr -o sas_pep.xvg На вопрос о выводе групп отвечаем ProteinЗависимость изменения гидрофобной гидрофильной поверхностей доступных растворителю от времени (красный - гидрофобная, зеленый - гидрофильная):
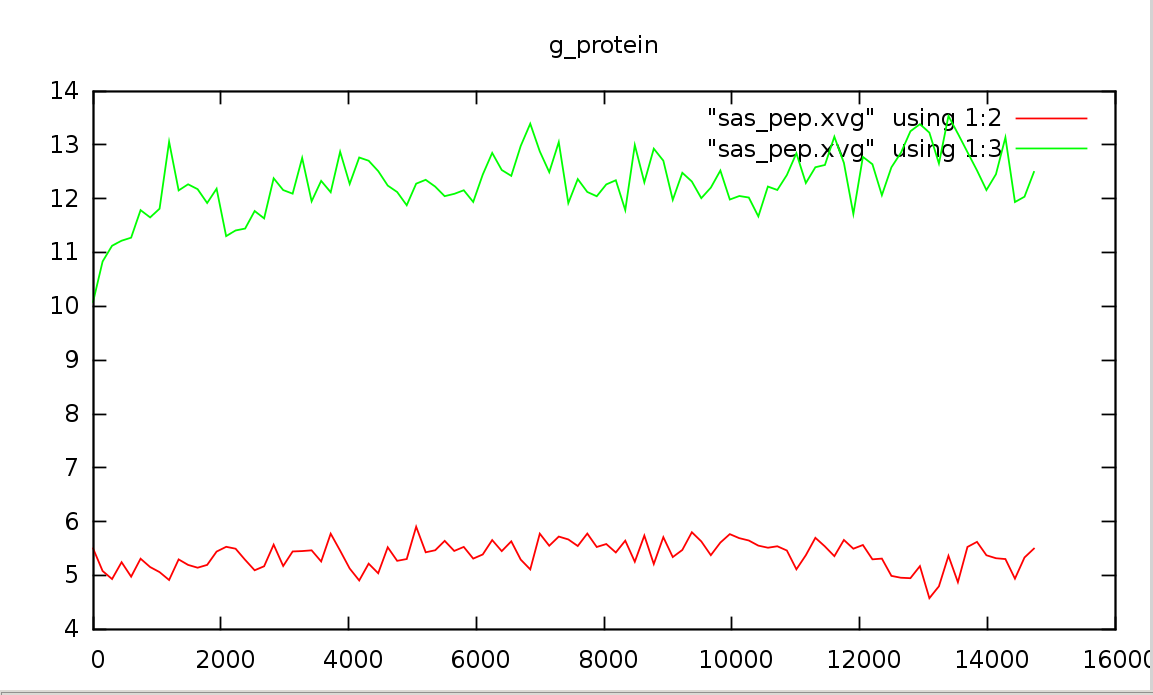
gnuplot> set title "g_protein"
gnuplot> plot "sas_pep.xvg" using 1:2 smooth csplines,\
>"sas_pep.xvg" using 1:3 smooth csplines
Возможные причины конформационного перехода:
Пептид наиболее расплавлен где-то в середине моделирования (см. п.1), на графике видно что, гидрофильная поверхность максимальна и контактирует с растворителем. Степень взаимодействия и гидрофильной, и гидрофобной практически не изменяется за все время моделирования.
Традиционным анализом является расчёт колчества образуемых водородных связей. Если мы будем исследовать связи между пептидом и пептидом, то это будут водородные связи в пептиде. Для конца траектории:
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep На вопрос о выводе групп отвечаем Protein, затем Faormamid (13)
Не менее интересно будет изучить количество вдородных связей пептид-формамид:
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep_sl На вопрос о выводе групп отвечаем Protein
Зависимость изменения гидрофобной гидрофильной поверхностей доступных растворителю от времени:
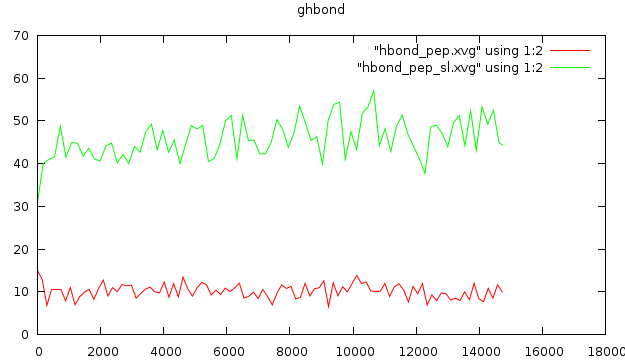
По каноническим представлениям, при плавлении должно происходить уменьшение количества водородных связей, но здесь этого не видно.
Конформационный переход также теоретически должен изменять количество водородных связей с растворителем, но это также не коррелирует с графиком.
export DSSP=/home/golovin/progs/bin/dsspcmbi
do_dssp -f pep_md.xtc -s pep_md.tpr -o ss
# Для просмотра переведём xpm в eps
xpm2ps -f ss.xpm -o ss.eps -by 10
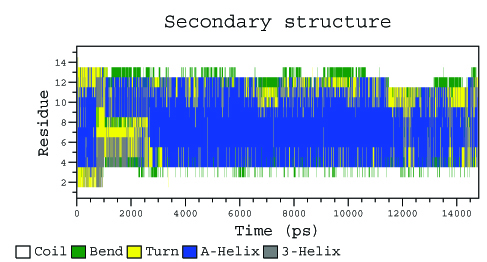
Как и в п.1, на диаграмме видно, что в середине моделирования концевые участки пептида расплавились из альфа-спирали. Больше всего, как и в п.1, меняется С-конец.