координаты пептида, 2xl1.pdb.
файл с ячейкой уравновешеных молекул формамида, fam_em.gro.
файл дополнительной топологии для формамида, fam.itp.
файл праметров для минимизации энергии em.mdp.
файл праметров для "утряски" воды pr.mdp.
файл праметров для молекулярной динамики md.mdp.
Их надо скачать в рабочую директорию.
cd lynx/md
pdb2gmx -f 2xl1.pdb -o pep -p pep -ff amber99sb -water tip3p
editconf -f pep.gro -o pep_ec -d 1.5
grompp -f em -c pep_ec -p pep -o pep_em -maxwarn 1
mdrun -deffnm pep_em -v
Изменение максимальной силы в ходе оптимизации геометрии:
До:
F-max = 4.37039e+03 on atom 146 F-Norm = 2.00120e+03После:
Low-Memory BFGS Minimizer converged to machine precision in 96 steps, but did not reach the requested Fmax < 1. Оптимизировать о значения Fmax <1 не получилось. Potential Energy = 9.5273560e+02 Maximum force = 3.2095328e+02 on atom 30 Norm of force = 8.4225098e+01
genbox -cp pep_em -p pep -cs fam_em.gro -o pep_s
В выводе программы написано количество добавленных молекул формамида:
Added 902 molecules Generated solvent containing 5412 atoms in 902 residues
; Include forcefield parameters
добавим #include "fam.itp":
; Include forcefield parameters
#include "fam.itp"
Добавим количество молекул формамида в запись [ molecules ]. Было:
[ molecules ] ; Compound #mols pep 1стало:
[ molecules ] ; Compound #mols Protein 1 FAM 902где 902 - количество молекул формамида.
System has non-zero total charge: -9.999999e-01(примерно -1, значит, нужно добавить 1 протон, чтобы нейтрализовать систему).
grompp -f em -p pep -c pep_s
genion -s pep_s -o pep_si -p pep -np 1
где 1 - это количество положительных ионов необходимых для нейтрализации заряда системы.
grompp -f pr -c pep_si -p pep -o pep_pr -maxwarn 1
mdrun -deffnm pep_pr -v
editconf -f pep_pr.gro -o pep_pr.pdb
editconf -f pep_si.gro -o pep_si.pdb
Визуализация в PyMol изменений в системах:
До утряски молекулы растворителя упорядочены:
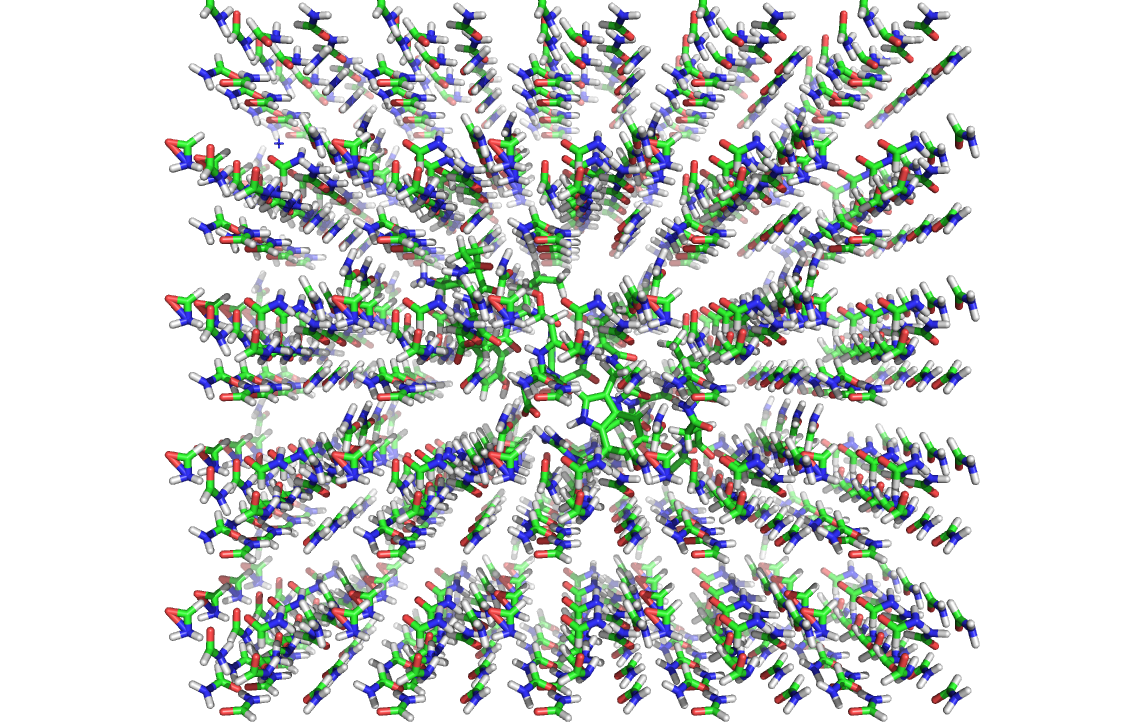
После утряски:

cd ..
scp -r md/* skif:fbb/lynx/
ssh skif
cd fbb/Ivanov
grompp -f md -c pep_pr -p pep -o pep_md -maxwarn 1
mpirun -np 16 -maxtime 5 -q test /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Просмотреть ход счёта можно в файле mdrun_mpi.out-....
less mdrun_mpi.out-....
Надо нажать shift+. для перехода в конец файла.
При отсутствии ошибок переходим к основному моделированию.
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Номер в очереди ID=155736.
Ориентировочное время счёта 10 часов.