export PATH=${PATH}:/home/preps/golovin/progs/bin
export MOPAC_LICENSE=/home/preps/golovin/progs/bin
Оптимизация порфирина.
Аннотация порфирина в виде SMILES:c1cc2cc3ccc(cc4ccc(cc5ccc(cc1n2)[nH]5)n4)[nH]3
файл 1.smi
С помощью obgen, была построена 3D структура порфирина:
obgen 1.smi > 1.mol
Полученная структура была просмотрена в PyMol:
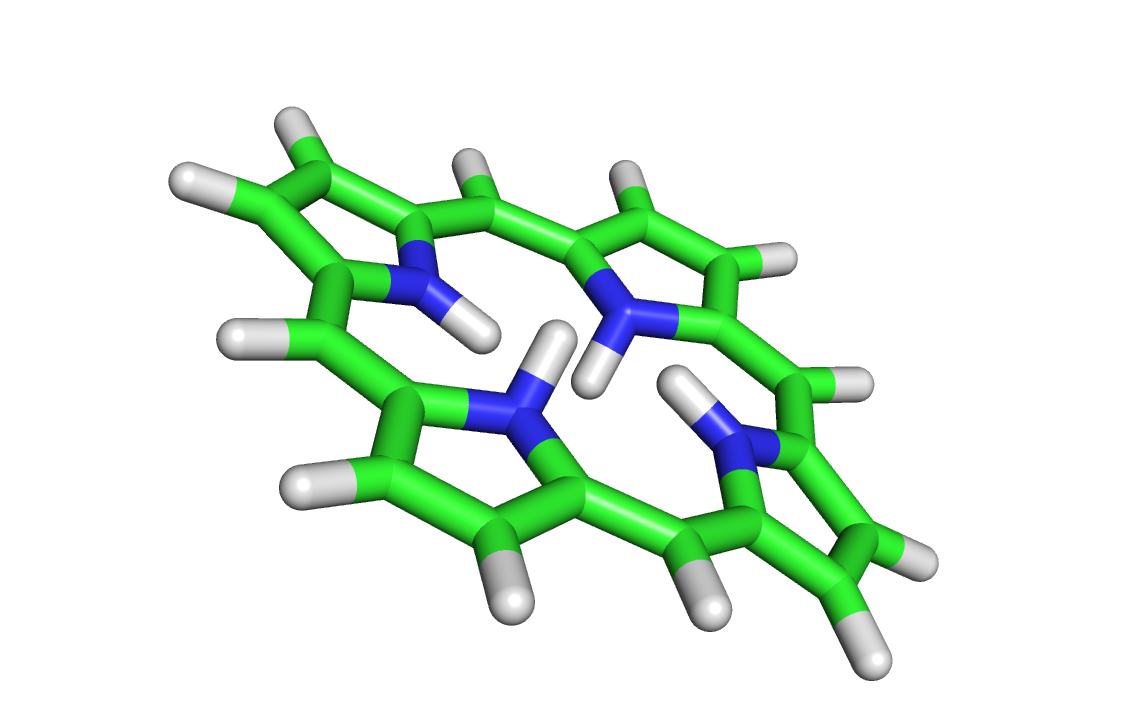
2 водорода лишние, ненужные водороды были удалены ( atom->remove atoms):
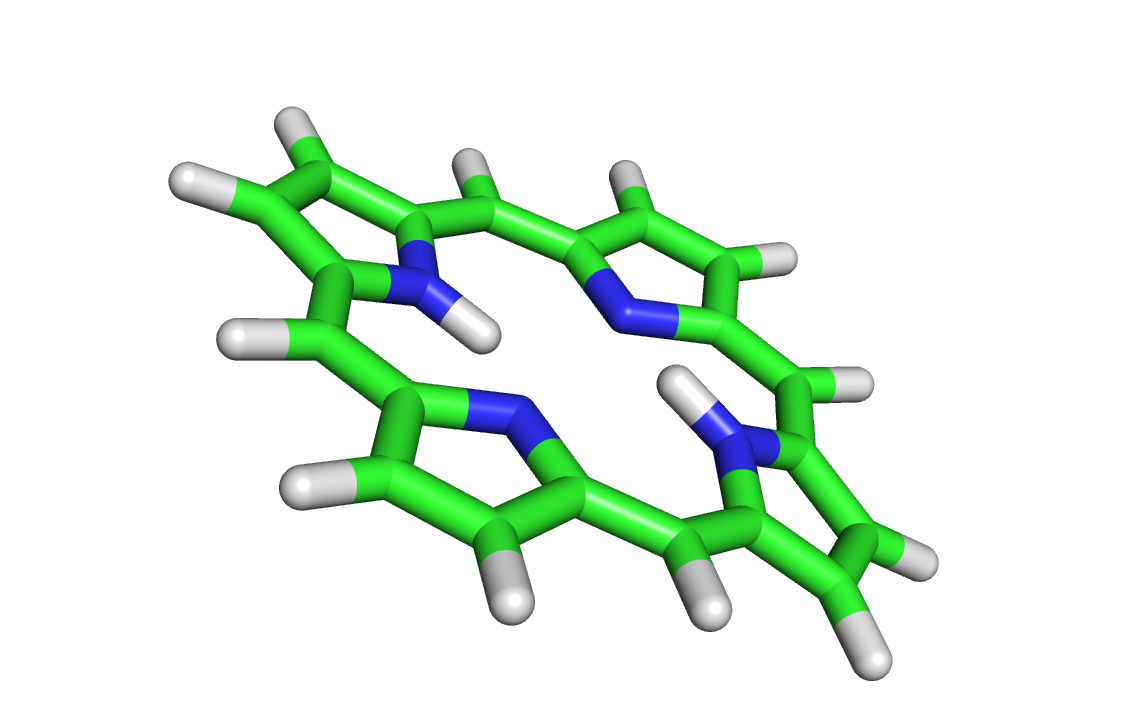
см. pdb файл
С помощью babel координаты в mol формате были переформатированы во входной файл для Mopac:
babel -ipdb porf.pdb -omop 1_opt.mop -xk "PM6" - c помощью -xk мы задаем параметризацию типа pm6.
Полученный файл был обработан с помощью программы Mopac:
MOPAC2009.exe 1_opt.mop
Выходной файл: 1_opt.out. Для сравнения он был переформатирован в pdb: babel -imopout 1_opt.out -opdb 1_opt.pdb
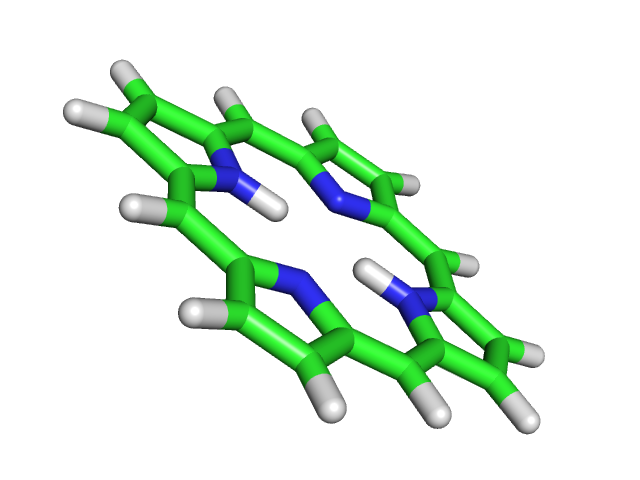
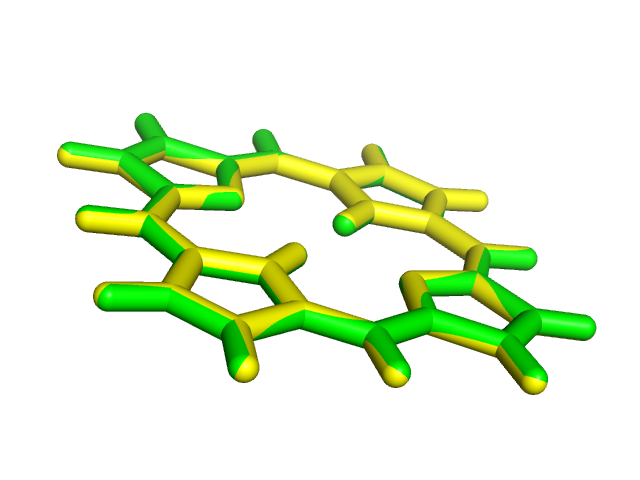
Полученная структура плоская, в отличие от исходной.
Аналогичным образом была проведена оптимизация с параметризацией AM1:
babel -ipdb porf.pdb -omop 2_opt.mop -xk "AM1"
MOPAC2009.exe 2_opt.mop
babel -imopout 2_opt.out -opdb 2_opt.pdb
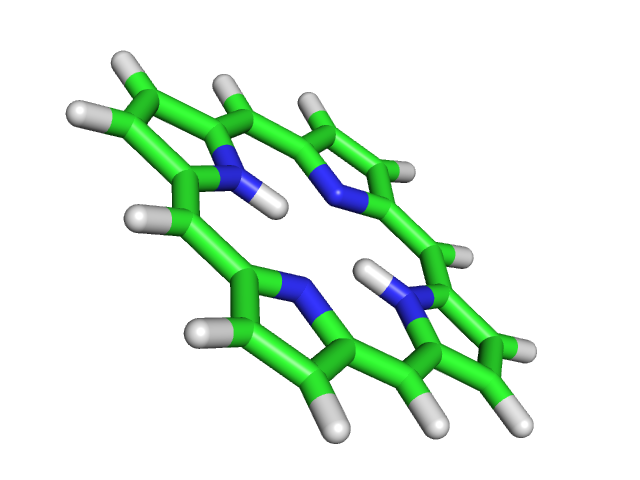 pdb
pdb Полученная струтура тоже плоская. Разницы между результатами этих двух оптимизаций нет, как видно из наложения структур:
Возбужденные состояния порфирина и спектр поглощения молекулы.
Для расчёта возбуждённых состояний была создана копия mop файла из предыдущего пункта: 1_opt_spectr.mop. Для указания Mopac о необходимости расчёта возбуждённого состояния в конец файла было добавлено:пустая строка
cis c.i.=4 meci oldgeo
some description
Выдача MOPAC ( MOPAC2009.exe 1_opt_spectr.mop): 1_opt_spectr.out. Значения энергий для электронных переходов указаны в конце файла:
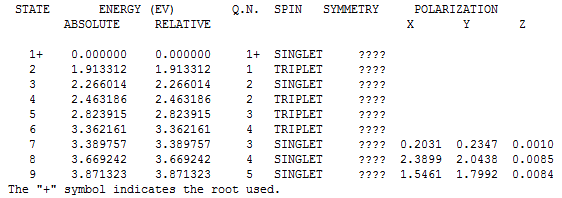
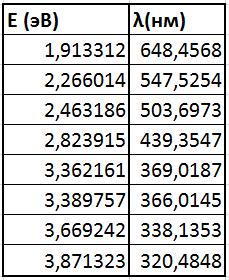
На основании значений энергий переходов и формулы E=(hc)/λ были рассчитаны длины волн, при которых происходят эти переходы:
Оптимизация 1,4-бензохинона (парабензохинона).
Для молекулы 1,4-бензохинона (SMILE: O=C1C=CC(=O)C=C1) была определена геометрия как с помощью obgen (benz.mol), так и Мopac (benz.pdb): obgen benz.smi > benz.molbabel -ipdb benz.pdb -omop benz_opt.mop -xk "PM6"
MOPAC2009.exe benz_opt.mop
babel -imopout benz_opt.out -opdb benz_opt.pdb
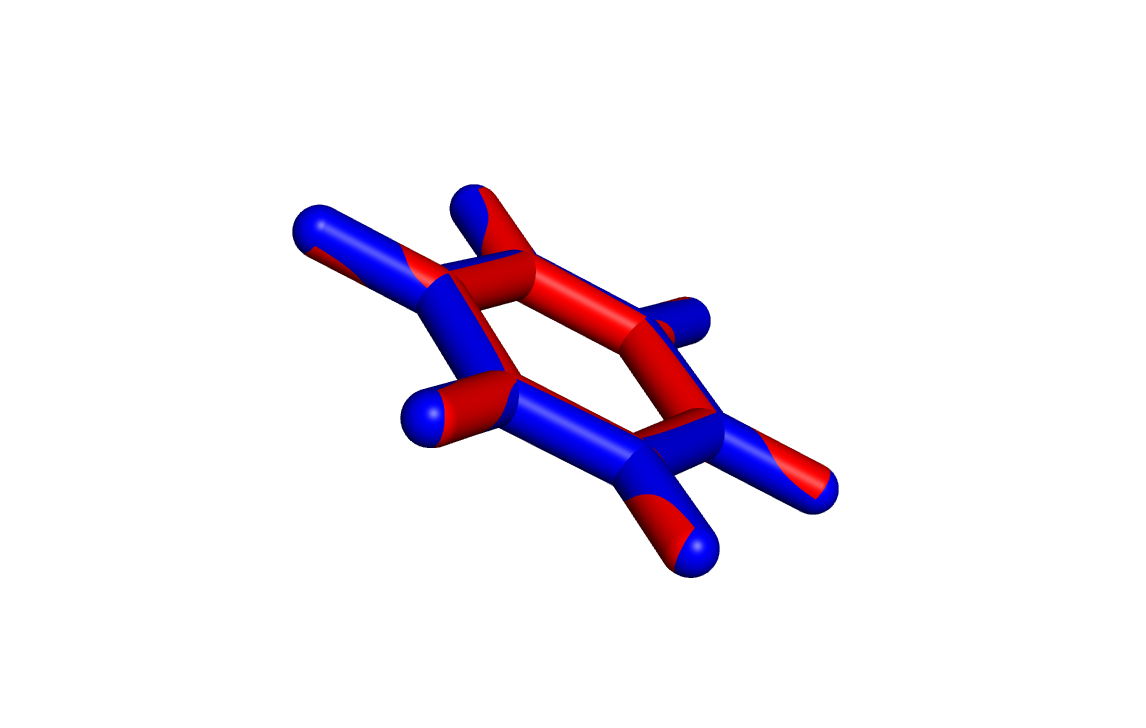
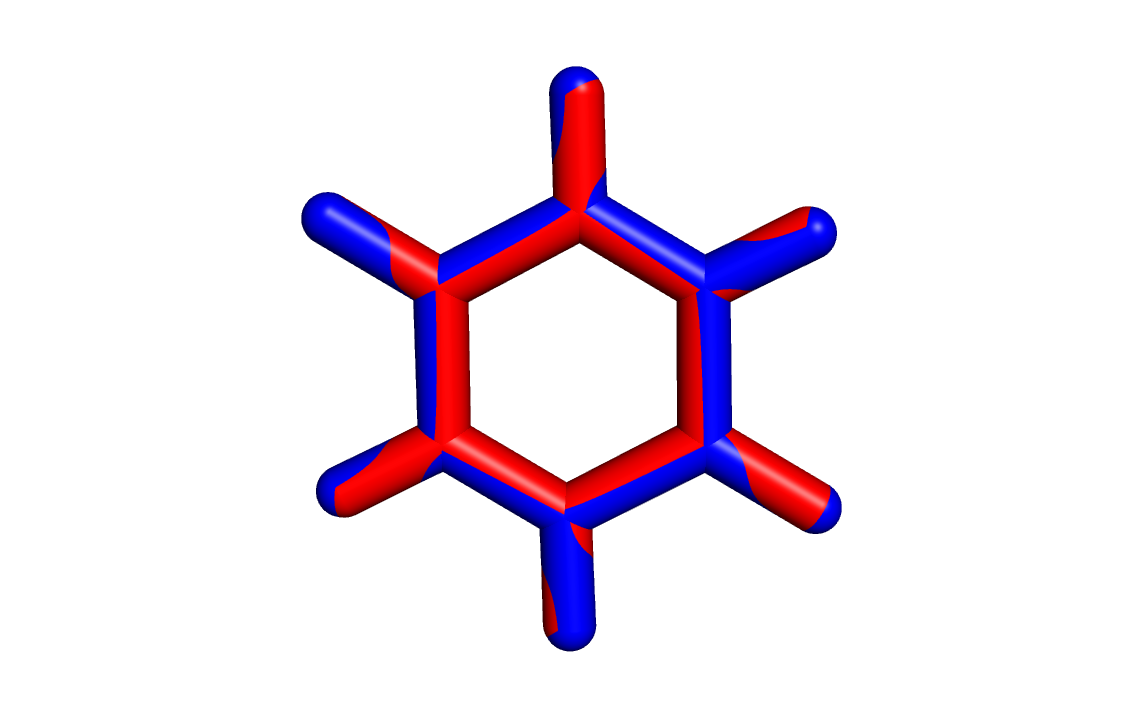
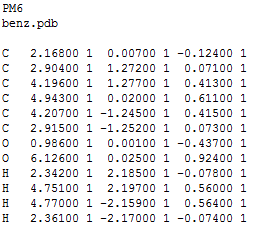
Геометрия, полученная obgen, выделена красным, а Mopac - синим. Из картинки заметно, что связи C-C, рассчитанные в obgen, немного короче этих же связей, рассчитанных в Mopac. Также была определена геометрия дианиона этой молекулы. Для этого в первую строчку mop файла было добавлено слово CHARGE=-2, затем явным способом было указано на каких атомах должен находиться отрицательный заряд (см. файл):
было
 стало
стало 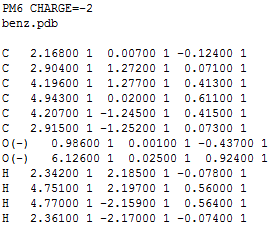
MOPAC2009.exe benz_opt_modified.mop
babel -imopout benz_opt_modified.out -opdb b_opt_m.pdb
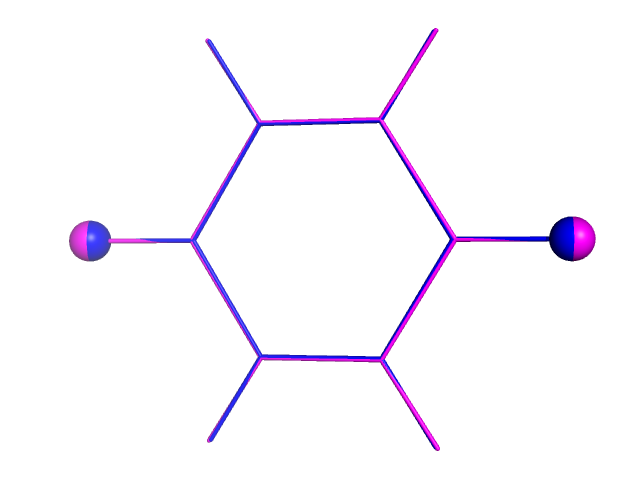
Как видно, связь C-O в дианионе длиннее, чем в нейтральной молекуле (magenta - дианион, синий - исходная структура).
Тиминовые димеры
Дан димер.1) Проведена оптимизация геометрии этого димера, при заряде системы 0.
2) Проведена оптимизация результата из пункта 1) , при заряде системы +2.
3) Проведена оптимизация результата из пункта 2), при заряде системы 0.
Энергии всех трех состояний (1, 2, 3)
I состояние: TOTAL ENERGY = -3273.58217 EV
II состояние: TOTAL ENERGY = -3253.90755 EV
III состояние: TOTAL ENERGY = -3273.69464 EV
Возбуждённое состояние при добавлении электронов не перешло обратно в исходно состояние, потому что переход в начальное состояние менее энергетически выгоден.