-
1. Построение дерева по нуклеотидным последовательностям.
- C помощью команды entret sw:rs2_bacan stdout | less получаю запись Swiss-Prot с описанием моего белка и вывожу её на экран.
- Введя /EMBL нахожу имя записи описывающей полный геном бактерии.
- entret embl:AE016879 stdout | less - вывожу запись EMBL с полным геномом бактерии на экран
- seqret embl:AE016879[9335:10841] - получаю нужную последовательность (ген кодирующий 16S РНК данной бактерии)
- seqret embl:AE015927[8715:10223] -sreverse stdout >>clote.fasta - получение комплементарной последовательности.
- Получение нужных нуклеотидных последовательностей (алгоритм привожу для первой из моего списка бактерии Bacillus anthracis):
Результат выравнивания
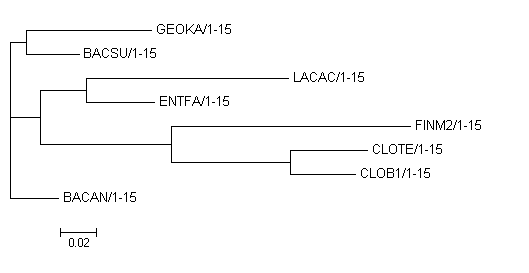
Для построения дерева использовал программу fdnaml и получил
((GEOKA/1-15:0.06932,BACSU/1-15:0.02895):0.00912,((LACAC/1-15:0.11207,ENTFA/1-15:0.03789):0.02522,(FINM2/1-15:0.13230,(CLOTE/1-15:0.04276,CLOB1/1-15:0.03623):0.06580):0.07255):0.01690,BACAN/1-15:0.02669);
неукорененное дерево
если укоренить:
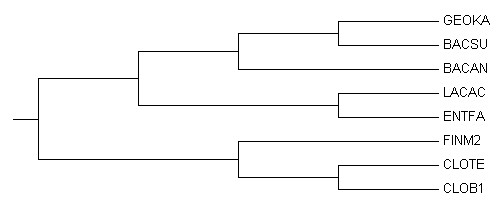
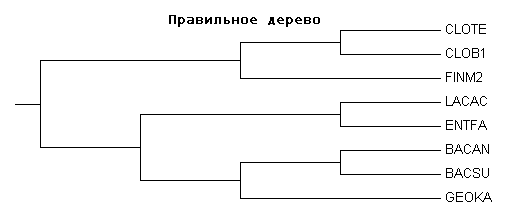
Дерево очень близко к правильному,
но присутствует ветвь {GEOKA,BACSU}vs{BACAN,CLOTE,CLOB1,ENTFA,LACAC,FINM2} внутри ветви {GEOKA,BACSU,BACAN}vs{CLOTE,CLOB1,ENTFA,LACAC,FINM2}
в правильном дереве есть ветвь {BACSU,BACAN}vs{GEOKA,CLOTE,CLOB1,ENTFA,LACAC,FINM2}
и гораздо лучше построенных по белковым последовательностям. Поэтому можно предположить, что как минимум для близких таксонов использование нуклеотидных последовательностей дает лучший результат, чем использование аминокислотных.
-
2. Построение и анализ дерева, содержащего паралоги.
Результат выравнивания
Было построено дерево с помощью JalView по алгоритму neighbour joining using % identity
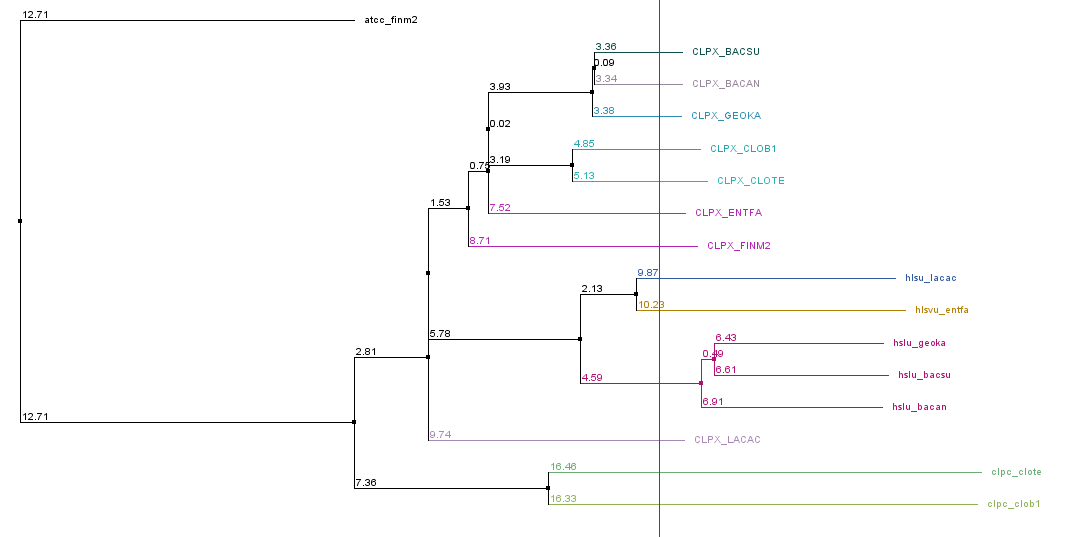
Примеры ортологов: HSLU_GEOKA, HSLU_BACAN, HSLU_BACSU и CLPX_GEOKA, CLPX_BACAN, CLPX_BACSU
Примеры парологов: CLPX_BACSU с HSLU_BACSU и CLPX_BACAN с HSLU_BACAN