Домены и профили
Построение HMM-профиля семейства белков и проверка его работы
Для выполнения данного практикума был выбран домен Cas_Cas2CT1978 (PF09707) из базы данных Pfam. Данный домен представляет собой минорную ветвь семейства Cas2 CRISPR-ассоциированных белков. Информация о домене:
- Число белков в seed: 70
- Число белков в full: 795 (файл)
- Число белков Uniprot: 3155
- Длина профиля домена: 86

В разделе Domain organisation содержится 2 архитектуры, одна из которой является двудоменной. Второй домен – рибонуклеаза T RNase_T (PF00929). Данный домен в последовательностях белков следует после домена Cas2. Данной архитектурой обладают 65 последовательностей (файл с идентификаторами белков с архитектурой).
Распределение длин белков с доменом представлено на рис. 1. Белки можно разделить на две группы, большая часть белков имеет длину около 100 аминокислот, это как раз белки, относящиеся ко второму типу архитектуры (содержащие только домен Cas2). К меньшей группе относятся белки длины около 300, входящие в рассматриваемую двухдоменную архитектуру.

С помощью программы MUSCLE было получено выравнивание белков, затем в ходе ревизии были удалены две слишком схожие последовательности (Remove redundancy 95%) и был удален небольшой участок перед первым доменом, который был у некоторых последовательностей. В результате получился следующий файл.
Следующий этап – построение HMM профиля и поиск по профилю. Для этого использовались следующие команды:
hmm2build 3_HMM twodom_al.fasta —> создание профиля. Длина – 304 hmm2calibrate 3_HMM —> калибровка hmm2search -E 0.1 --cpu 1 3_HMM full.fasta > hmmrsearch.txt —> поиск по профилю, файл с результатом. Всего нашлась 661 последовательность.
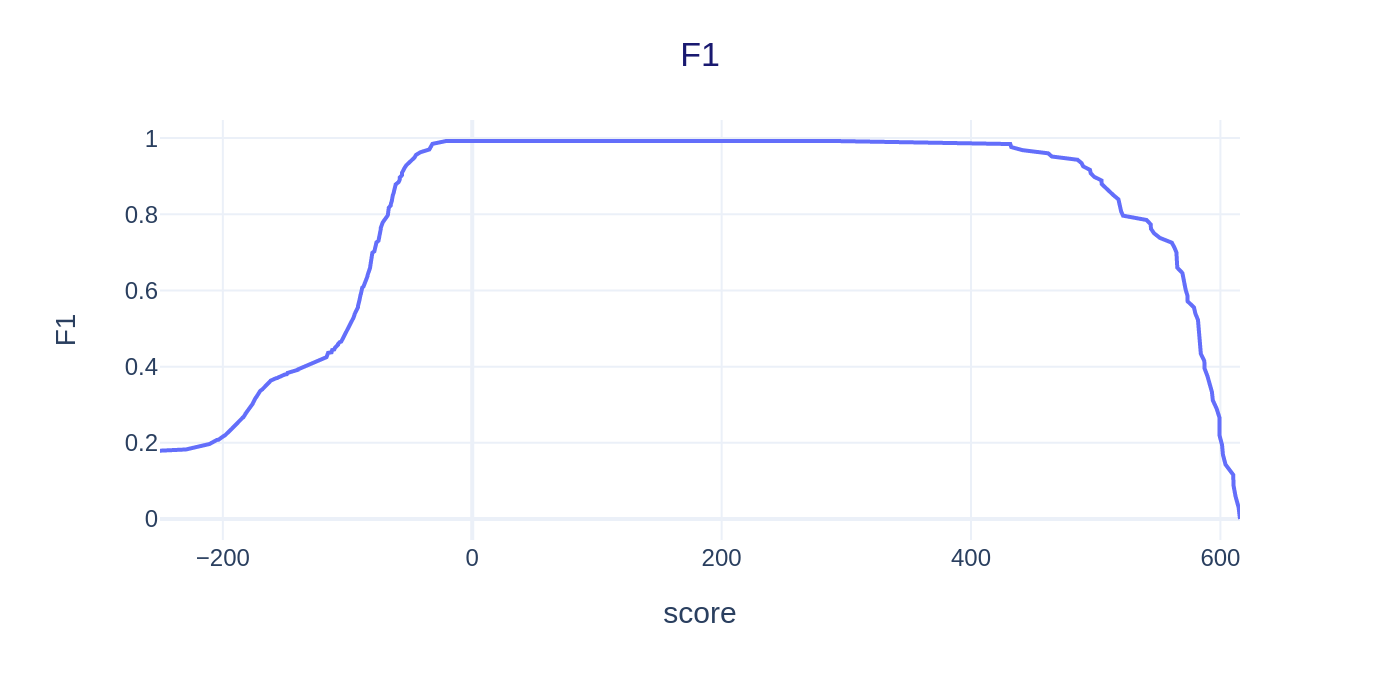
Далее проверим правильность работы профиля. Для этого создадим таблицу, содержащую информацию о находках, взятую из выдачи hmm2search, и добавим туда столбец True, где yes означает то, что найденный белок находится в списке белков с архитектурой, и no в противном случае. Как мы видим, нашлись все 65 последовательностей с соответствующей профилю доменной архитектурой. Далее, используя информацию из колонки True и формулы из презентации вычисляем sensitivity, 1 - specifity и F1 для каждой находки. По этим данным можно определить порог веса, удовлетворяющий нашим требованием для чувствительности и специфичности предсказания принадлежности той или иной находки к семейству.
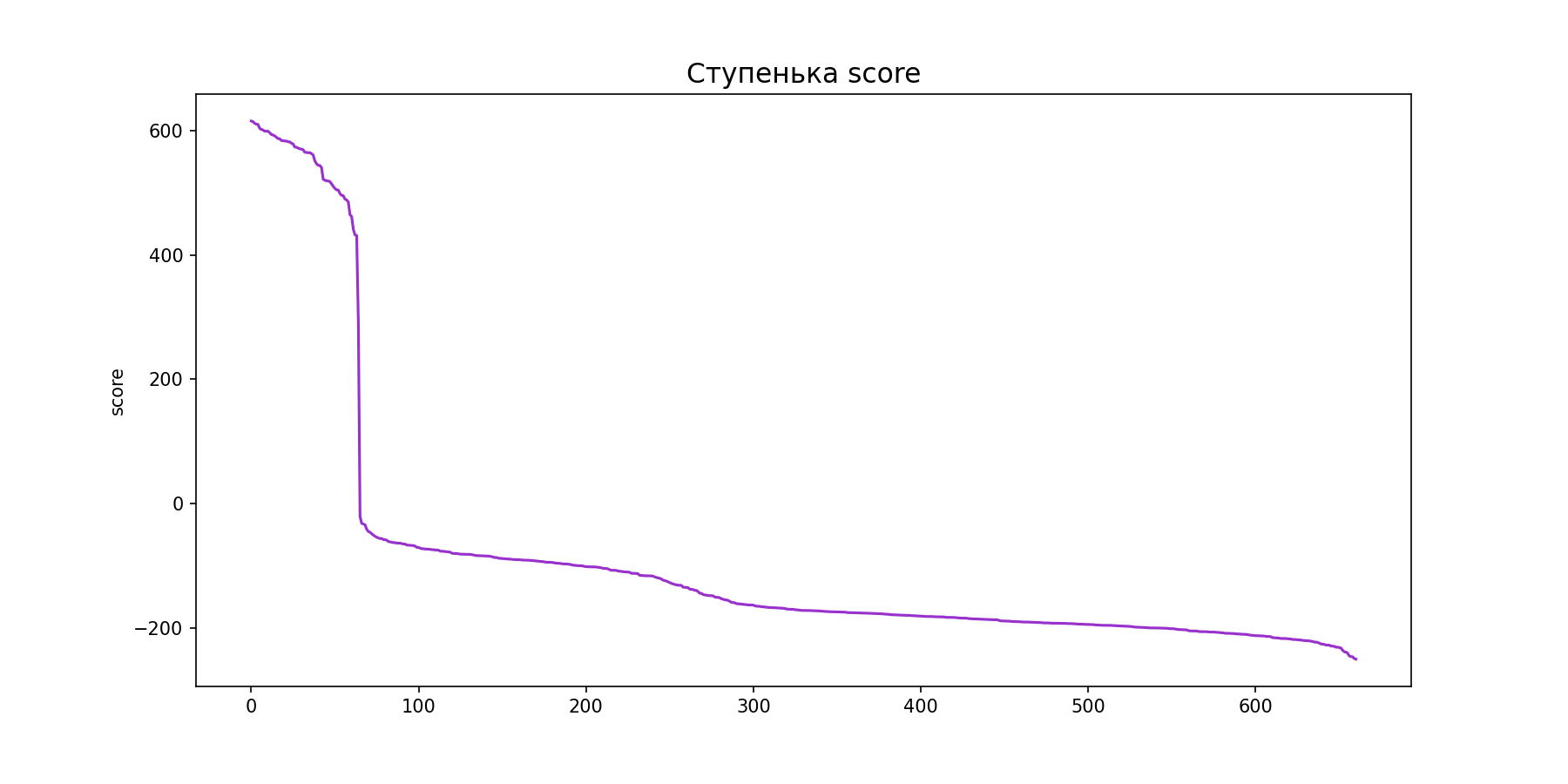
Для начала посмотрим на распределение весов находок, осортированных по убыванию веса. Видим так называемую ступеньку – место резкого падения значений. Данный спад наблюдается в районе 65 находке, ее вес равен 291.6. Вес 66 находки равен уже -20.9. Все находки до 65-й (т.е. находки с лучшим весом) относятся к выбранной двудоменной архитектуре, поскольку в колонке True для них стоит yes.
Построим ROC-кривую для опрделения порога веса. Полученный график представлен на рисунке 3. Данный график не подходит для определения порога, потому что все находки с наиболее высоким score принадлежат к одной архитектуре, что создает резкий скачок при переходе к находкам с более низкими весами. Можно попробовать построить график среднего гармонического чувствительности и специфичности с целью нахождения порога. С ростом веса среднее гармоническое постепенно увеличивается, затем выходит на плато в районе значений score от -20.9 до 431.5, затем постепенно уменьшается. График F1 показал схожие результаты с графиком распределения весов. Вероятно, из-за слишком резкого изменения параметра score данные графики не дадут никакой полезной информации для точного определения порога предсказания, такова специфика данной задачи. По моему мнению, в качестве порогового веса лучше всего подойдет вес 65 находки – 291, это граничный вес между последовательностями с двухдоменной архитектурой и без нее.