Протонирование и автоматизация поиска контактов
Задание 1
Для выполнения задания была структура скиталидо-глутаминовой пептидазы из Scytalidium lignicola (аскомицет) в комплексе с ингибитором (2IFW). Белок представлен в виде гомодимера.
В задании использовался сервис PDB2PQR. Его встроенный алгоритм PROPKA рассчитывает возможность влияния электростатики от полярных участков молекулы белка на протонирование титруемых химических групп. Сервису было задано значение pH = 5.5, соответствующее условиям кристаллизации. Все остальные параметры оставлены по умолчанию. После завершения анализа были получены файлы форматов .log и .pqr.
В файле .log содержится таблица с предсказанными значениями pKa остатков. Были рассмотрены такие аминокислотные остатки Asp и Glu, чьи pKa выше pH кристаллизации (равный 5.5).
Были выбраны два разных остатка с наибольшими pKa:
Asp57 (цепь B) - 9.37;
Glu3 (цепь B) - 11.83.
Далее эти остатки были рассмотрены в структуре.
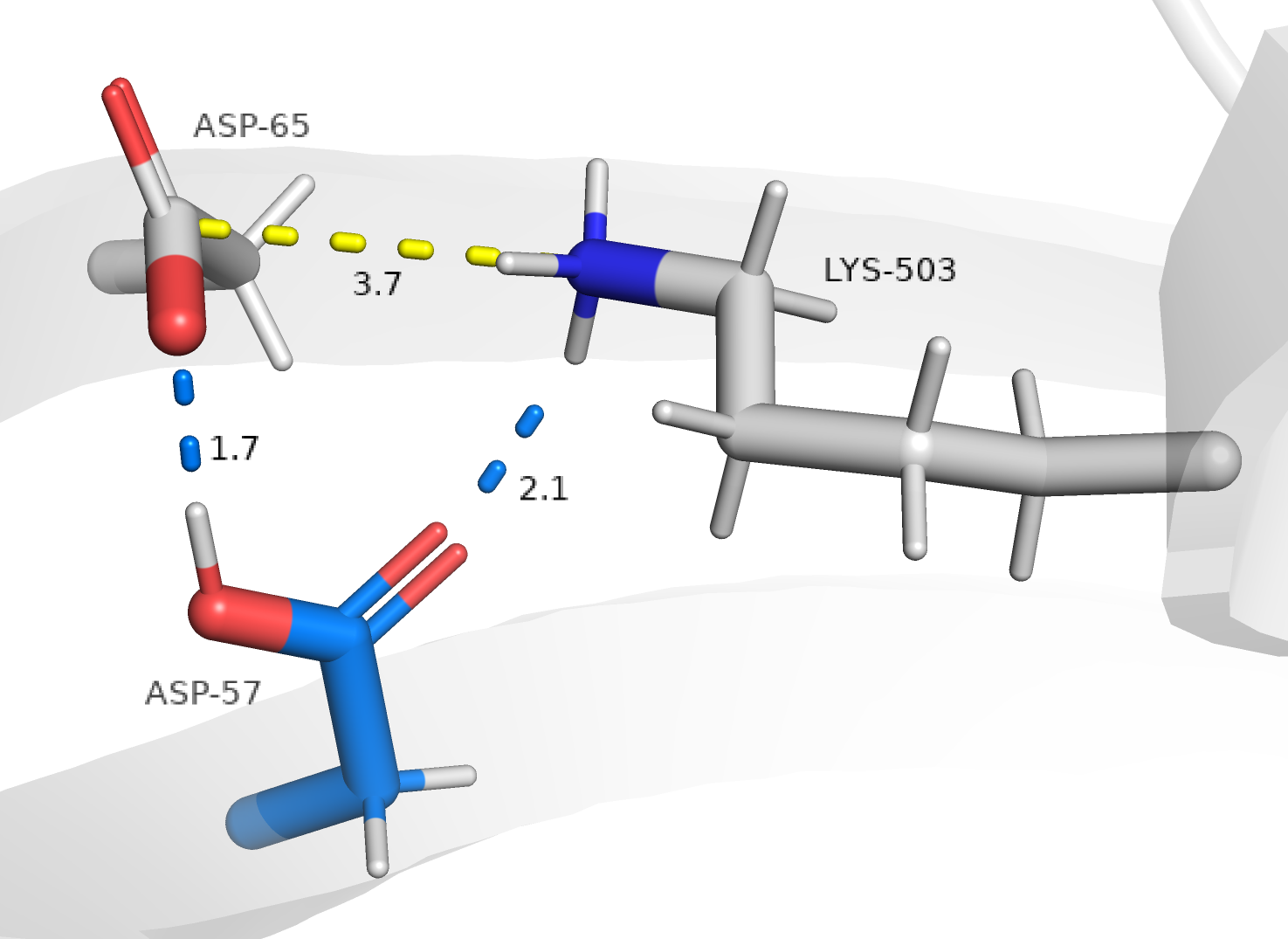
ASP57 (рис. 1)
Остаток Asp57 протонирован, а рядом расположенный Asp65 - нет, и, соответственно, он является отрицательно заряженным. Если оба остатка имели бы отрицательный заряд, возникло бы их электростатическое отталкивание. Однако протонирование Asp57 предотвращает такую ситуацию и позволяет образовать водородную связь между остатками. Кроме того, рядом располагается положительно заряженный лизин, способный образовывать солевой мостик с отрицательно заряженным Asp65. Это способствует стабилизации структуры белка.
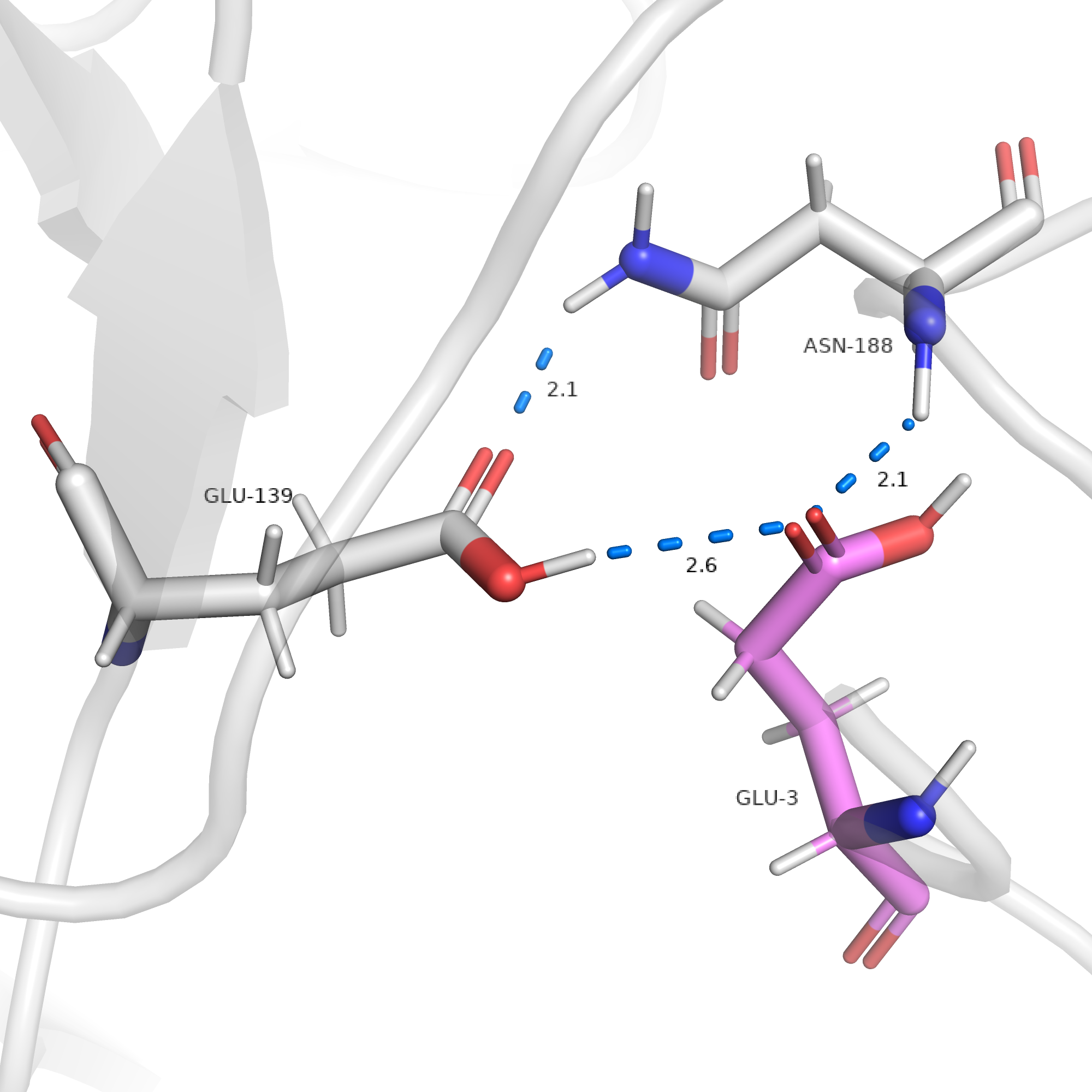
GLU3 (рис. 2)
Протонирование Glu3 устраняет его отрицательный заряд, что предотвращает электростатическое отталкивание с соседним остатком глутамата (Glu139). Однако никаких новых взаимодействий протон не дает. Глутамат-сосед образует водородную связь с остатком аспарагина, что стабилизирует структуру. Однако протонирование Glu3 приводит к сближению водорода у боковой цепи Glu3 и главной цепи Asn188, и это создает не очень неблагоприятное стерическое взаимодействие между ними.
Задание 2
В следующем задании была выдана структура белкового филамента 6SIH, бактериального флаггелярного кэп-белка FliD, который расположен на кончике жгутика и отвечающий за его ротацию. Структура получена с помощью электронной микроскопии.
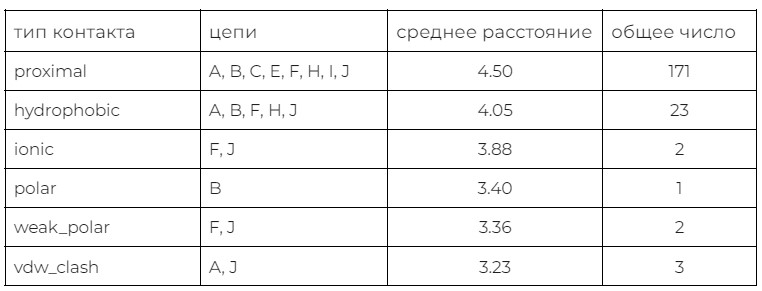
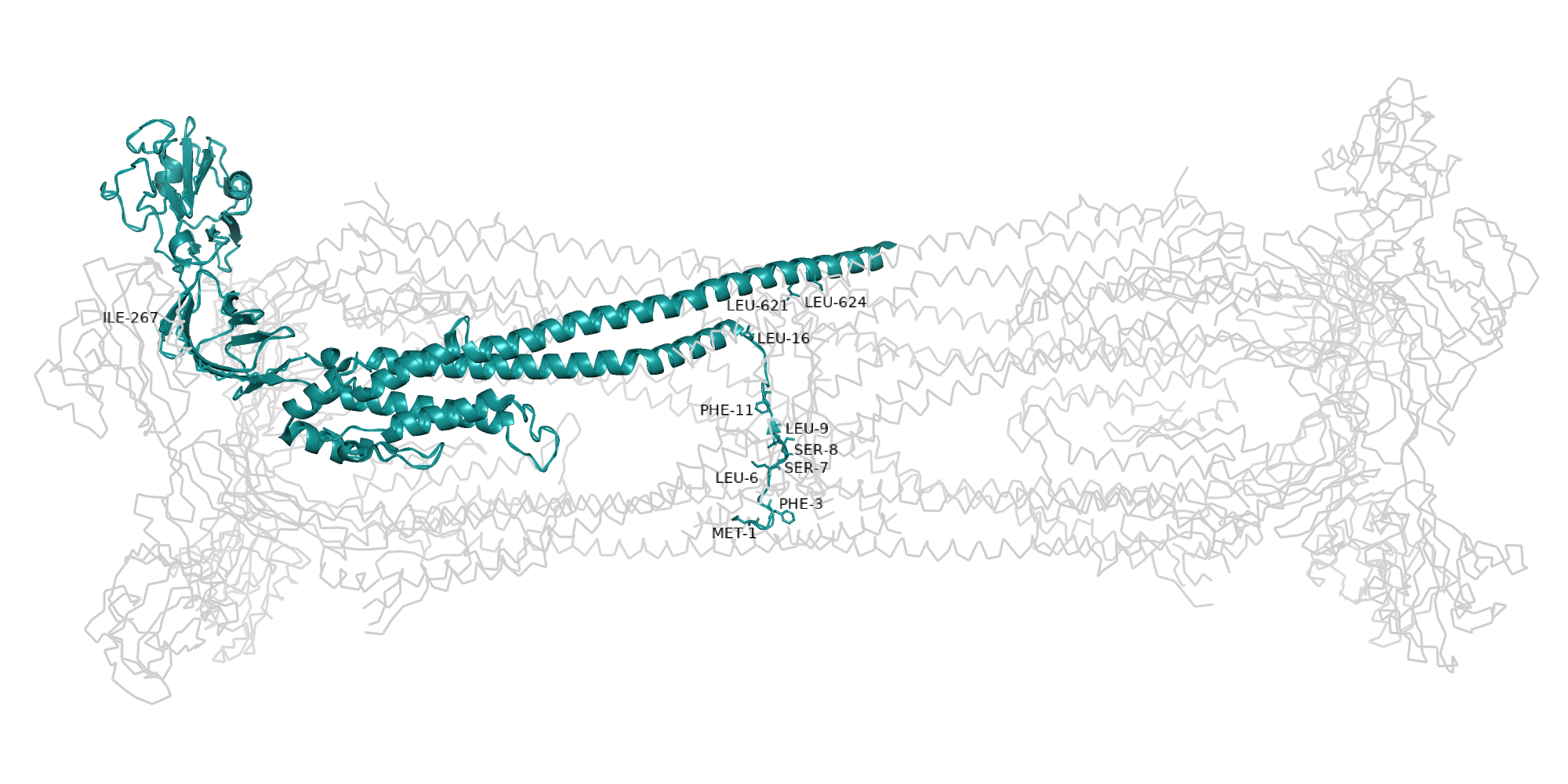
Структура состоит из 10 мономеров. Для анализа контактов была выбрана цепь G (рис. 3). С помощью выданного ноутбука, позволяющего искать контакты одной цепи с соседними (Arpeggio), было выяснено, что цепь взаимодействует с 8 соседями (цепи A, B, C, E, F, H, I, J; все, кроме цепи D) (см. таблицу ниже).
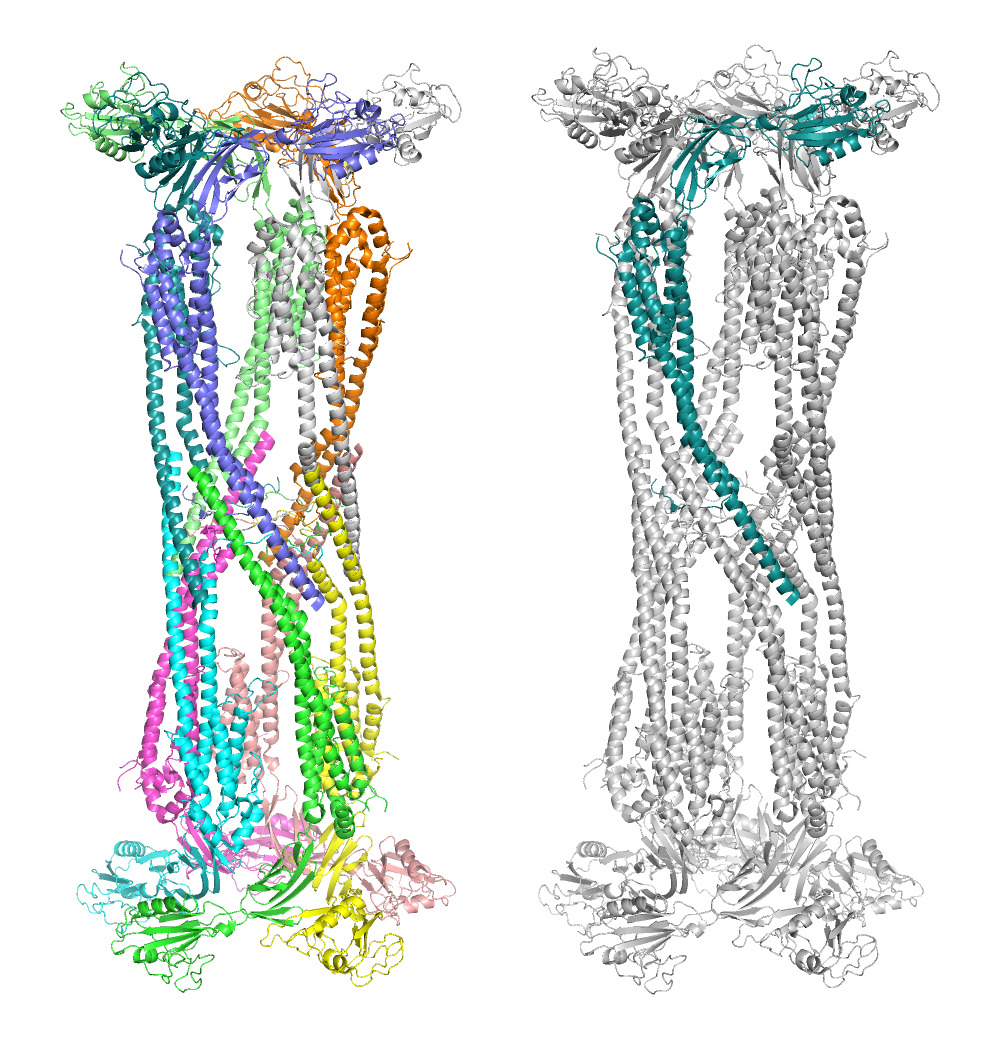
Самые часто встречающиеся контакты - проксимальные, далее - гидрофобные, остальные связи найдены в единичных экземплярах. Самое большое число контактов цепь G образует с цепью A (58), J (50) и F (44), меньше всего с I (6) и E (2).
Самое большое число контактов образуют аминокислоты:
• Ser7 - 15
• Ser8 - 15
• Phe11 - 14
• Leu9 - 14
• Leu6 - 13
• Met1 - 12
• Phe3 - 12
• Leu16 - 11
• Leu624 - 10
• Leu621 - 10
• Ile267 - 10
Очень много аминокислот, образующих гидрофобные взаимодействия (все, кроме серина), что можно связать с большим количеством найденных гидрофобных контактов. Локализация остатков на рис. 4.
Ссылка на сессию PyMOL здесь.