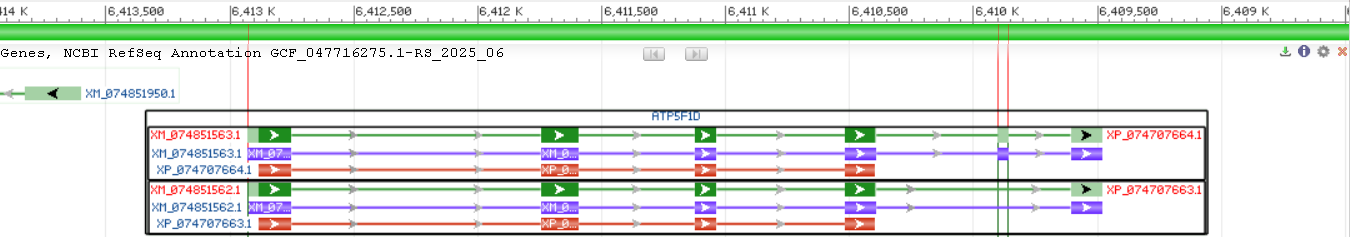
В таблице белковых последовательностей для сборки генома Длиннохвостой неясыти (лат. Strix uralensis) было найдено две записи, соответствующих дельта субъединице АТФ синтазы F (id: XP_074707663.1, с которым проводилась дальнейшая работа и id: XP_074707664.1). Соответствующие этим записям аминокислотные последовательности абсолютно идентичны (см. сопроводительные материалы 1), но получены с отличающихся вариантов транскрипта одного гена ATP5F1D (GeneID: 141935415, см. рис. 1, последовательность CDS и прилегающих к ней участков см. в сопроводительных материалах 2). Данный ген относится к нуклеотидной записи с идентификатором NC_133998.
Чтобы выяснить, насколько последовательность белок-кодирующей области изучаемого гена консервативна, был проведён поиск с помощью BLAST методами blastn и tblastn (чтобы сравнить результаты работы двух методов) данной последовательности среди геномов Пауков (лат. Araneae) из базы данных refseq genomes. Данный таксон был выбран, т.к. он удалён от исследуемого организма и обнаружение в нём схожих последовательностей было бы показательно.
В базе данных refseq_genomes хранится 4 сборки, относящихся к таксону Пауки (4 сборки генома четырёх различных видов пауков).
Метод blastn был выбран для поиска нуклеотидных последовательностей, хоть сколько-либо похожих на исследуемую, среди последовательностей геномов пауков из refseq_genomes (megablast не использовался, т.к. выбранные таксоны сильно удалены друг от друга и вероятность того, что найдутся в достаточной степени схожие последовательности, была не так высока). Был установлен размер слова 7, цены наличия и расширения гэпов, соответственно, 5 и 2 (см. рис.2).
Метод tblastn был выбран для поиска нуклеотидных последовательностей, гомологичных изучаемой, в сборках из refseq_genomes, принадлежащих паукам. Это возможно благодаря наличию у гомологичных белков достаточно продолжительных схожих участков. Т.к. одна аминокислота может кодироваться разными кодонами, то нуклеотидные последовательности, кодирующие гомологичные белки, могут отличаться так сильно, что не будут найдены методом blastn из-за особенностей работы BLAST. Но tblastn позволяет найти такие нуклеотидные последовательности за счёт их транслирования и сравнения с последовательностью белка (в данном случае альтернативой было использование tblastx, но tblastn был выбран из-за его большей скорости и из-за того, что tblastx мог найти негомологичные короткие рамки считывания). Установлена длина слова 2, для остальных параметров установлены значения по умолчанию (см. рис.3).
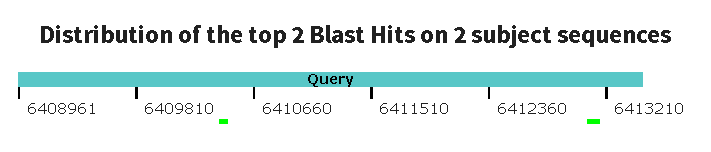
Примечание к рисункам 2 и 3: синим цветом выделены выравнивания со счётом 40-50 битов, зелёным - 50-80 битов, розовым - 80-200 битов.
Для поиска гомологов генов рРНК в геноме S. uralensis на основе её геномных последовательностей была создана локальная база данных 'owl' для blastn. Для этого использовалась следующая команда:
В файлы 16S_Ecoli.fasta.txt и 23S_Ecoli.fasta.txt были записаны последовательности, соответственно, 16S и 23S рРНК E. coli. Эти последовательности средствами локального Blast по очереди были выравнены с геномными последовательностями S. uralensis (т.е. с последовательностями из созданной базы owl). Использованные команды:
blastn -task blastn -db owl -query 16S_Ecoli.fasta.txt -out 16s_res.out -outfmt 7
blastn -task blastn -db owl -query 23S_Ecoli.fasta.txt -out 23s_res.out -outfmt 7
Для данной задачи был выбран алгоритм blastn, т.к. изучаемые последовательности консервативны и похожие участки в них можно найти даже у удалённых таксономически организмов.
Результаты blastn выравнивания 16S рРНК E. coli
Результаты blastn выравнивания 23S рРНК E. coli
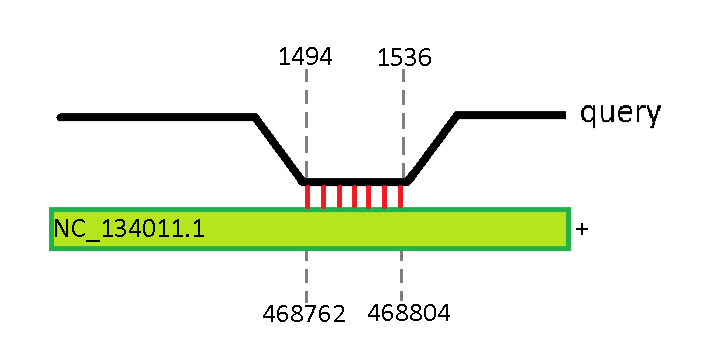
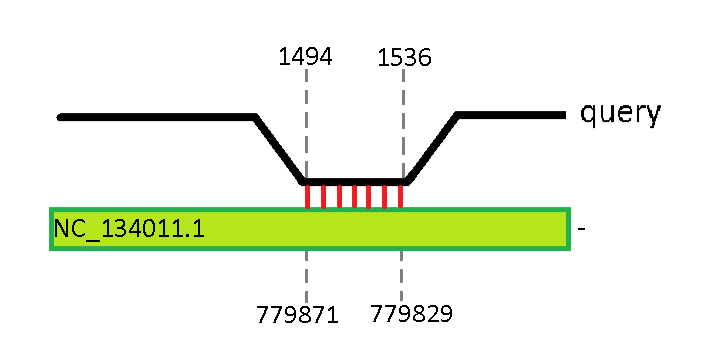
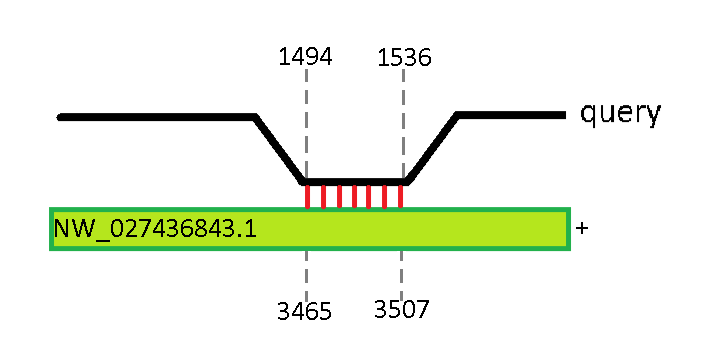
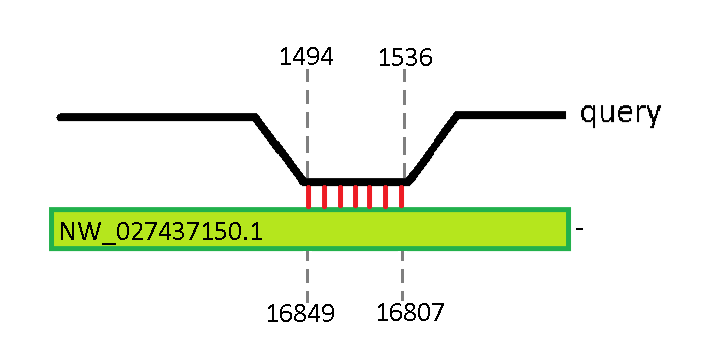
При выравнивании 16S рРНК E. coli было обнаружено 49 совпадений, из которых E-value менее 10^-2 имело 22 находки. Все они соответствуют участку 1494 - 1536 рРНК E. coli. Схемы, иллюстрирующие некоторые из этих находок, представлены на рис. 4 - 7.
При выравнивании 23S рРНК E. coli было обнаружено 110 хитов, из которых E-value менее 10^-2 имело 100 находок. На скэффолде NW_027436843.1 расположен один гомолог, содержащий участки 198-290 (участок 1), 1898-1983 (2), 2233-2300 (3) и 2442-2613 (4). На скэффолде NW_027436843.1 было обнаружено 8 гомологов, содержащих те же 4 участка. На прямой цепи 40 хромосомы обнаружено 4 гомолога, содержащих 3 и 4 участки, и 5 гомологов, содержащих все 4 участка. На обратной цепи той же хромосомы - 9 гомологов, содержащих все 4 участка.
Для проведения исследования были взяты последовательности хромосом представителей одного рода: архей Halorarum salinum (код доступа последовательности в GenBank: CP058579.1), некоторые особенности генома которой были описаны мной ранее, и Halorarum halophilum (GenBank AC: CP058529.1). Последовательнность хромосомы H. salinum была получена по ссылке в записи Dataset Genome изученной ранее геномной сборки археи на последовательность в базе данных nucleotide. Последовательнность хромосомы H. halophilum получена тем же способом (для поиска геномной сборки археи использовался поиск в Datasets Taxonomy).
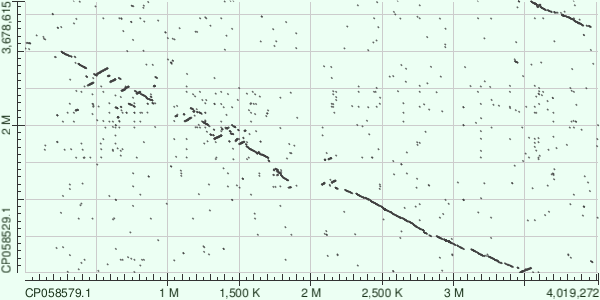
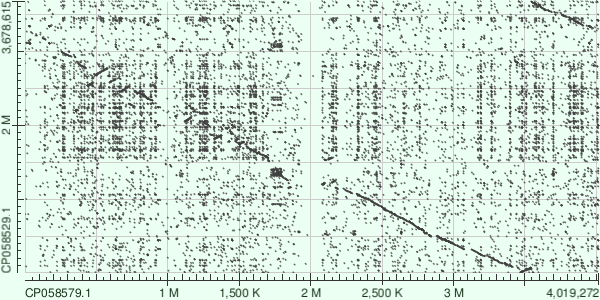
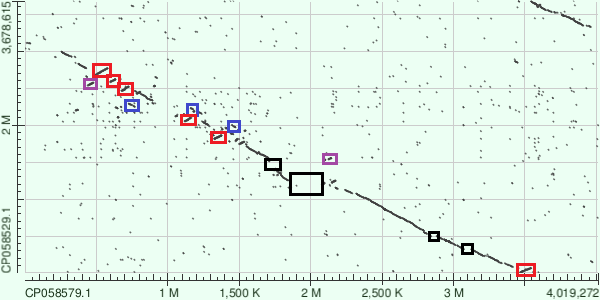
Полученные последовательности были выравнены друг с другом алгоритмами blastn и megablast (см. рис. 8 - 10). Как видим, алгоритм megablast обнаружил значительно меньше сходств, чем blastn. Это связано с тем, что megablast выравнивает лишь очень похожие последовательности. Также была произведена попытка применить алгоритм tblastx для выравнивания, однако это привело к выдаче ошибки. Вероятно, это связано с избыточным количеством выравниваний для разных рамок считывания.