Введение в анализ данных NGS (часть 2)
Картирование чтений на референсный геном
Картирование чтений на референсный геном осуществлено с помощью команды bwa mem, образец:
bwa mem -t 24 ../chr7.fna op1 op2 > 1.sam 2> bwamemstderr.log
Единственная использованная опция - подключение всех 24 ядер calc (-t 24), предварительно с помощью команды "top" была проверена загруженнооть CPU, чтобы не помешать другим процессам. В качестве референса был использован проиндексированный в предыдущем практикуме chr7.fna, в качестве триммированных чтений - парные op1, op2, вывод stdout перенаправлен в файл 1.sam, а stderr в bwamemstderr.
Описание SAM-файла
SAM расшифровывается как Sequence Alignment/Map формат. Это текстовый файл, использующий в качесте разделителя TAB, состоящий из секции заголовка (опциональной) и секции выравнивания. Если заголовок присутствует, он должен находиться раньше выравнивания. Строки заголовка начинаются с "@", а строки выравнивания - нет. Каждая строка выравнивания имеет 11 обязательных для ключевой информации о выравнивании, такой как позиция картирования, и переменного количества опциональных полей для специфичной информации.
SAM-файл состоит 11 основных столбцов и переменного количества опциональных:
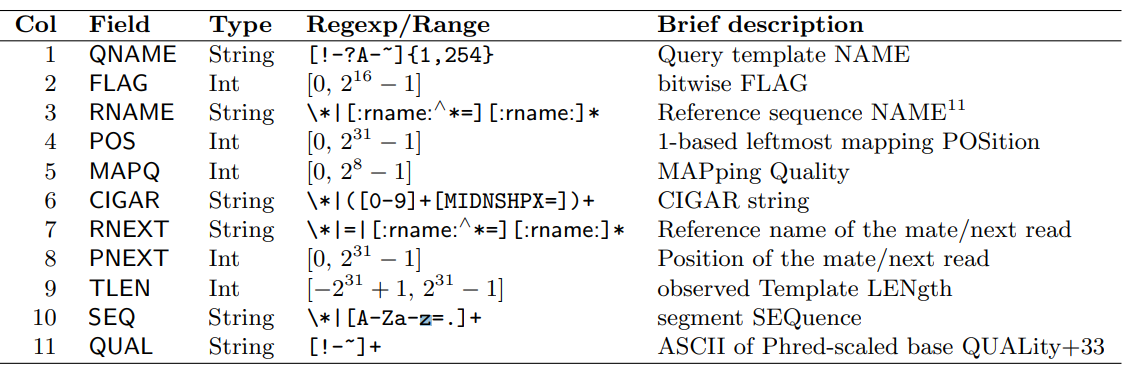
- QNAME: наименование шаблона запроса. Риды/сегменты с одинаковым QNAME принимаются как происходящие из одного шаблона. Символ '*' в поле показывет, что информация недоступна. В SAM-файле рид может заниманить несколько строк выравнивания, когда выравнивание химерное или когда даны несколько картирований.
- FLAG: комбинация битовых последовательностей, обозначающих, например, остутствие картирований (0x4). Полный набор кодов представлен в мануале.
- RNAME: наименование референсной последовательности выравнивания.
- POS: номер относительно единицы самой левой позиции картирования первой операции CIGAR, которая "потребляет" референсное основание (1 - самая левая позиция выравнивания).
- MAPQ: качество картирования, равное -10log(10;P) | P - вероятность ошибки в позиции картирования.
- CIGAR: CIGAR-строка из операторов, обозначающих, например, инсертцию или делецию по отношению к референсу (I, D соответственно). Полный список представлен в мануале.
- RNEXT: наименование референсной последовательности первичного выравнивания следующего рида в шаблоне.
- PNEXT: позиция первичного выравнивания следующего рида в шаблоне относительно единицы (1 - самая левая позиция выравнивания).
- TLEN: обозначенная наблюдаемая длина шаблона.
- SEQ: последовательность сегмента.
- QUAL: ASCII-код основания + 33 (phred33).
- TAG: опциональное поле, строящееся в формате TAG:TYPE:VALUE, где тэг - двухсимвольная строка из диапазона /[A-Za-z][A-Za-z0-9]/. Список типов представлен на изображении 2.

Формат описан на основании мануала, действительный заголовок (вывод команды head) находится по ссылке
Размер полученного оценен с помощью комнады "ls -lh", он составляет примерно 16 гигабайт.
Получение и индексирование bam файла
Для конвертации была использована команда "samtools sort" с единственной опцией наименования выходного файла "-o", подействованная на SAM-файл:
samtools sort -o 1.bam 1.sam
По данным команды "ls -lh", полученный BAM-файл занимает примерно 4.7 ГБ.
Индексация BAM-файла производена с помощью команды "samtools index" со всеми параметрами по-умолчанию:
samtools index 1.bam
Анализ bam файла
Для конвертации BAM-файла из бинарного в текстовый была использована команда "sam flagstat", ее вывод был перенаправлен в файл 1.bam.txt:
samtools flagstat 1.bam > 1.bam.txt
Из этого файла известна доля картированных чтений (13.47%) и картированных чтений в корректно картированных парах чтений (10.47%). Числа отличаются потому что не все картированные чтения находятся близко и не направлены друг к другу.
Получение картированных чтений
Название хромосомы было получено из fna-файла, с его помощью был выделен SAM-файл данной хромосомы командой:
samtools view -h 1.bam NC_000007.14 > 1.chr7.sam
Но можно было воспользоваться и командой "samtools faidx", она подтверждает название хромосомы.
Для определения доли картированных чтений аналогично была использована команда "samtools flagstat":
samtools flagstat 1.chr7.sam > chr7f.txt
Из полученного файла взято, что количество картированных чтений возрасла с 13.47% до 88.58%, а картированных в корректно картированных парах - с 10.47% до 69.09%.
Для выполения команды в точности SAM-файл хромосомы был переведен в BAM, и после из него были получены картированные чтения с помощью команды "samtools view":
samtools sort -o 1.chr7.bam 1.chr7.sam
samtools view -f 0x2 -bS 1.chr7.bam > cflags.bam
На этот файл подействовали командной "samtools flagstat":
samtools flagstat cflags.bam > cflags.txt
Итоговый файл был прочтен less, и в нем уже процент картированных чтений и картированных чтений в корректно картированны парах составляют 100%.
Подготовка выравнивания к поиску вариантов
Картированный BAM-файл хромосомы был маркирован с помощью команды "picard MarkDuplicates":
picard MarkDuplicates -M metrix.file.txt -I cflags.bam -O marked.bam
Для просмотра BAM-файла была вновь использована команда "samtools flagstat":
samtools flagstat marked.bam > mflags.txt
В результате прочтения текстового файла были найдены 494616 дублированных чтений, при этом доля картированных чтений составила 100%.