Markov Ivan, Введение в анализ данных NGS (часть 4)
Подготовка референса
В качестве рефренса был взят FNA-файл chr7 из предыдущих заданий, его индексирование произвдено командой "hisat2-build":
hisat2-build chr7.fna chr7
В результате работы команды были получены 8 бинарных файлов индексирования с типовым названием "chr7.i.ht2", где i - натуральное число от 1 до 8.
Оценка качества чтений
Для анализа былj взят чтениe РНК с ID SRR2015719_1. Далее оно было проанализировано с помощью программы "fastqc".:
fastqc SRR2015719_1.fastq.gz
Из HTML-файла отчета по ссылке была представлена следцющая информация:
Общее количество чтений составило 34642046.
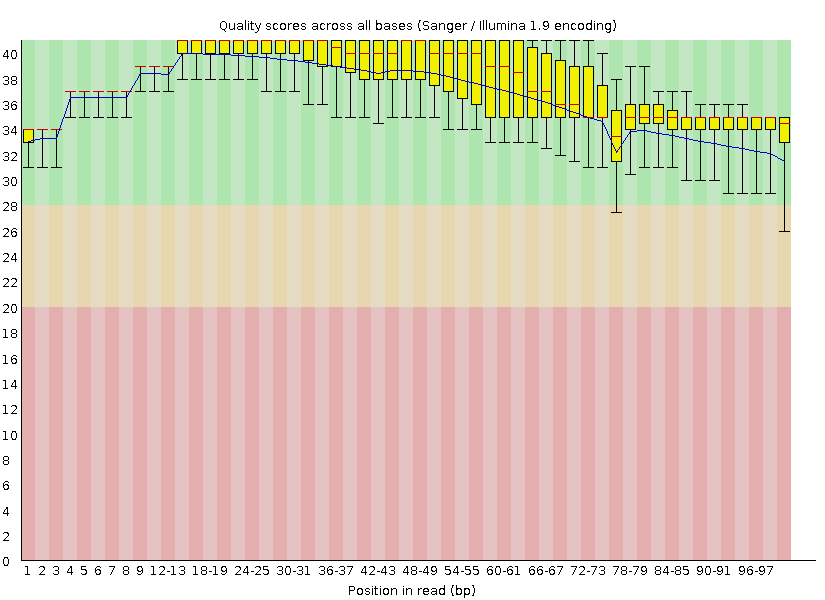
Качество чтений в целом хорошее, оно возрасатет по ломанной от 33 до 40, а затем по прямой падает до 34. Отклонение вниз 2-5 единиц, с небольшими исключениями (например последний цикл элонгации). Графически представлено на изображении 1
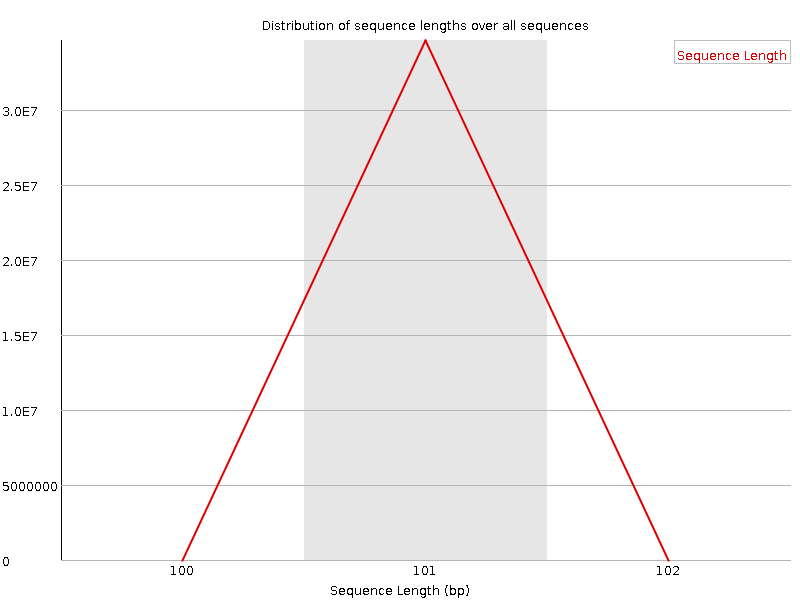
Длина чтений от 100 до 101 нуклеотида, крайне близкое число к обычному при секвенировании по технологии Illumina. Распределение представлено на изображении 2.
Картирование чтений на референс
Картирование произведено командой "hisat2" с параметрами: -x chr7 (обязательный, указывает на референсный геном), -k 3 (сообщать до 3 выравниваний на рид) и -U SRR2015719_1.fastq.gz (обязательный, указание на файл с неспаренными чтениями). Вывод программы пренаправлен в файл "1.sam", а stderr - в "hstx.log". Целиком команда выглядит как:
hisat2 -x ../chr7 -k 3 -U SRR2015719_1.fastq.gz > 1.sam 2>hstx.log
При рассмотрении лог-файла обнаружено, что все чтения (34642046) - непарные, 94.89% (32870335) выровнены 0 раз, 4.87% (1688545) выровнены единожды, а 0.24% (83166) выровнены более одного раза. Общая частота выравнивания - 5.11%.
SAM-файл конвертирован в BAM-файл с помощью команды "samtools sort":
samtools sort -o 1.bam 1.sam
Далее полученный файл был проиндексирован:
samtools index 1.bam
Чтения, которые легли на данную хромосому были получены командой:
samtools view -h 1.bam NC_000007.14 > 1.chr7.sam
Также был дополнительно создан бинарный файл картирования:
samtools sort -o 1.chr7.bam 1.chr7.sam
Поиск экспрессирующихся генов
Файл с разметкой генов имеет расширение .gtf и является текстовым файлом, поля которого разделены TAB. Ысего в этом файле 9 полей:
- seqname: названием хромосомы или скэффолда.
- source: название программы, сгенерировавшей эту feature, или источник данных.
- feature: название типа feature (например, Gene, Variation, Similarity).
- start: начальная позиция feature при условии, что последовательность начинается с 1.
- end: конечная позиция feature при условии, что последовательность начинается с 1.
- score: значение типа с плавающей точкой (float).
- strand: определяется как + или -.
- frame: принимает значения "0","1" или "2", показывает что первое основание feature является первым основанием кодона, аналогично "1" - вторым и.т.д.
- attribute: список разделенных полуколонкой тэг-значащих пар, предоставляющий дополнительную информацию о каждой feature.
Количество картированных чтений на каждый ген разметки посчитано с помощью команды "htseq-count". Использованные параметры: -f bam (формат файла картирования bam), -s no (дата из неспецифичного по странду образца), -m union (режим для пересечения feature, дефолтный) и -t exon (feature - экзон, дефолтное). В качестве файла чтений был взят 1.chr7.bam, а генов разметки - gencode.chr7.gtf. Итоговая команда:
htseq-count -f bam -s no -m union -t exon rna/1.chr7.bam gencode.chr7.gtf > gr.txt
Количетство чтений, попавших в границы генов оценивается командой:
wc -l gr.txt | head -n $lines-5 gr.txt | awk '{s+=$2}END{print s}'
Оно составляет 1229406. Чтения без feature указаны в конце файла gr.txt, их 384113. Просмотреть эту строку можно с помощью команды:
tail gr.txt