Введение в анализ данных NGS
Описание файла с референсом chr7.fna
Для рассмотрения использовался файл chr7.fna. Так как аббревиатура fna означает "fasta nucleic acid", в этом референсном файле содержится последовательность седьмой хромосомы человека в FASTA-формате. Образец из начала файла:
>NC_000007.14 Homo sapiens chromosome 7, GRCh38.p13 Primary Assembly NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
Индексация референса
Осуществлена с помощью команды:
bwa index -a bwtsw chr7.fna
Команда "bwa index" индексирует файл в FASTA-формате, опция "-a" задает алгоритм, в данном случае bwtsw служит для работы с более крупными базами данных, например цельным геномом человека, название файла показывает, к чему применена команда. Результатом работы стало появление пяти файлов с именами: chr7.fna.amb, chr7.fna.ann, chr7.fna.bwt chr7.fna.pac b chr7.fna.sa. Например, в аннотации содержится описание fasta-последовательности и, вероятно, количество нуклеотидов:
less chr7.fna.ann 159345973 1 11 0 NC_000007.14 Homo sapiens chromosome 7, GRCh38.p13 Primary Assembly 0 159345973 21 (END)
Описание образца с NCBI ID SRR10720412
Следующая информация взята с сайта NCBI.
- Сслыка на информацию об образце.
- Прибор - Illumina Genome Analyzer IIx.
- Организм - Homo sapiens.
- Стратегия отмечена как "OTHER", но в названии эксперимента написано, что это - секвенирование экзома.
- Чтения пароноконцевые (Layout: PAIRED).
- Ожидаемое количество чтений (spots) - 41398792.
Проверка качества исходных чтений
Для анализа использовались следующие команды:
fastqc SRR10720412_1.fastq.gz fastqc SRR10720412_2.fastq.gz
В итоге были получены два html-файла с результатами анализа, ссылка на них: SRR10720412_1_fastqc.html, SRR10720412_2_fastqc.html.
Количество пар чтений (Total sequences) совпадают у "прямых", "обратных" чтений, а также с указанным авторами, оно равно 41398792.
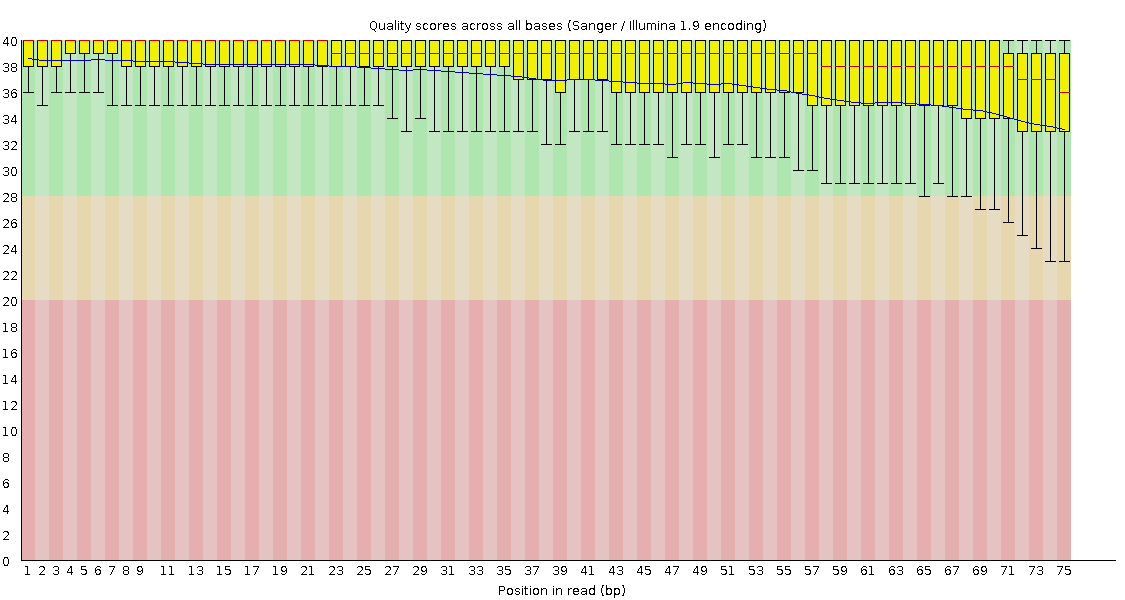
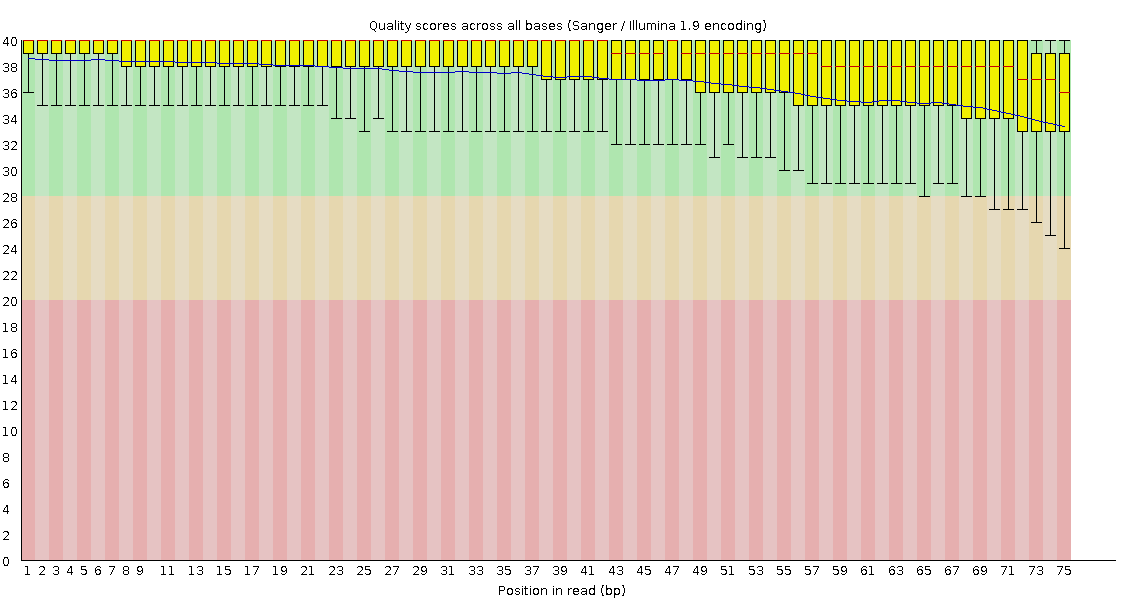
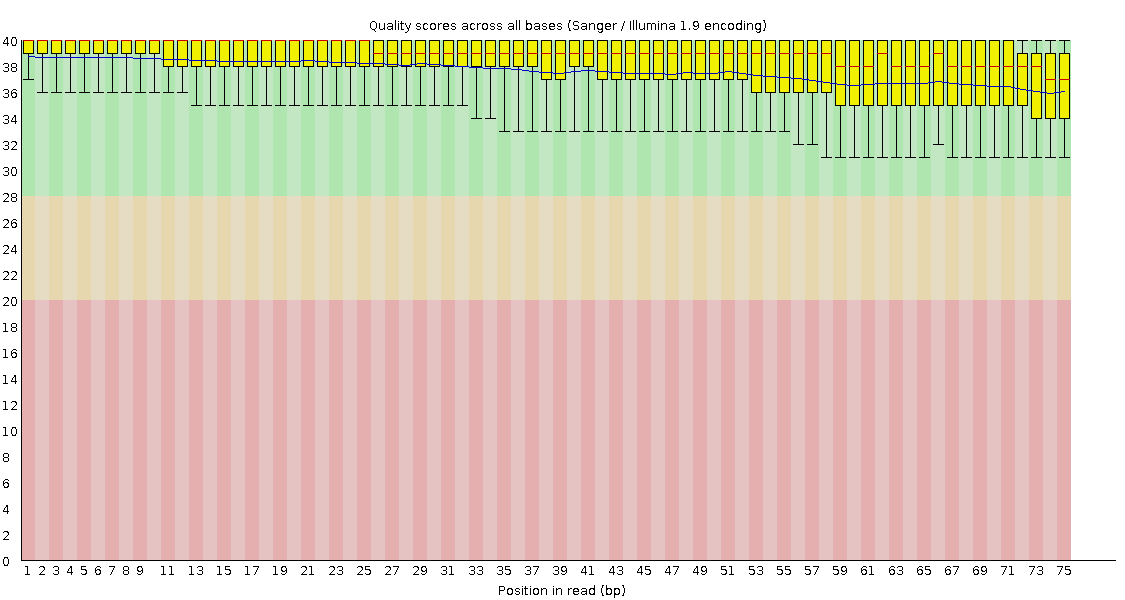
Качество чтений хорошее, от 40 с отклонением на -4, до 36 с отклонением на -10. Качество постепенно ухудшается с номером цикла элонгации. Подробные изображения представлены ниже.
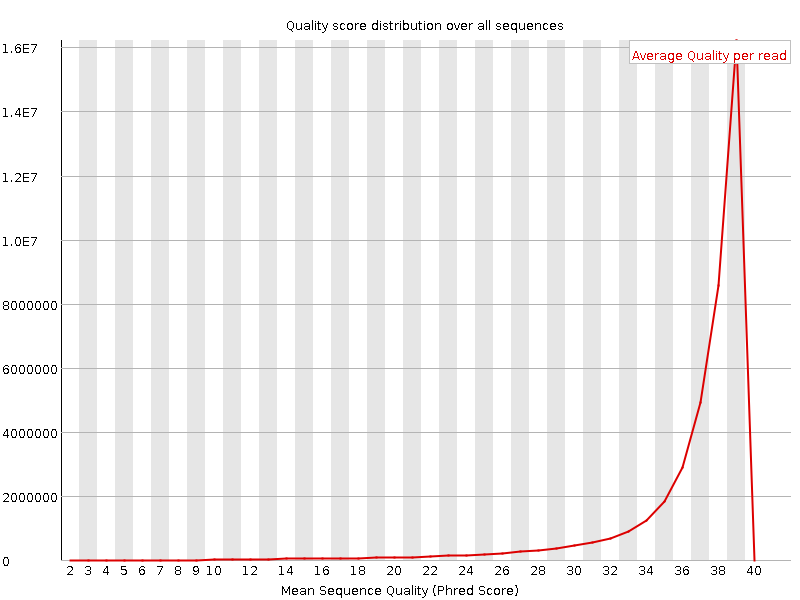
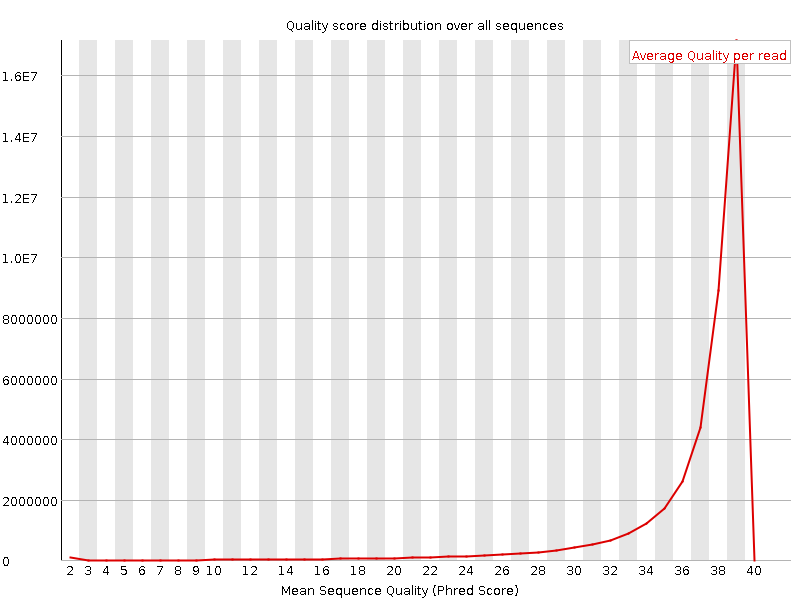
Распределение показало, что большинство участков секвенирования имеют хорошую точность. Чем меньше, точность, тем меньше участков ей обладают. Для "обратного" чтения заметен небольшой пик в окрестности точности 2, возможно, погрешность. Графики представлены на изображениях ниже.
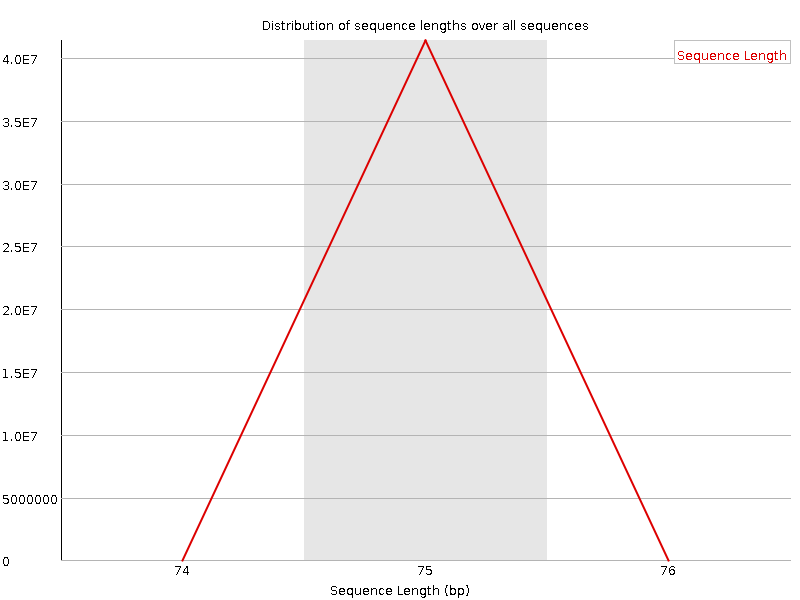
Средняя длина участка секвенируемой последовательности - 75 нуклеотидов. Это соответствует технологии Illumina, в которой используются небольшие участки, часто до 100 нуклеотидов. Резульаты представлены на рисунках ниже.
Фильтрация чтений
Для фильтрации чтений была использована программа "trimmomatic", использованная команда и его вывод:
java -jar /usr/share/java/trimmomatic.jar PE -phred33 -trimlog t12.log SRR10720412_1.fastq.gz SRR10720412_2.fastq.gz op1 ou1 op2 ou2 TRAILING:20 MINLEN:50 TrimmomaticPE: Started with arguments: -phred33 -trimlog t12.log SRR10720412_1.fastq.gz SRR10720412_2.fastq.gz op1 ou1 op2 ou2 TRAILING:20 MINLEN:50 Input Read Pairs: 41398792 Both Surviving: 39853177 (96.27%) Forward Only Surviving: 678011 (1.64%) Reverse Only Surviving: 659625 (1.59%) Dropped: 207979 (0.50%) TrimmomaticPE: Completed successfully
Команда запущена на java в режиме парных концов (PE) с качеством чтения phred33, сохранением log в файл t12.log (сслыку установить не получается, файл слишком велик для копирования в домашнюю директорию на kodomo), исходными файлами с названиями чтений, выходными файлами op1-2, ou1-2 (output paired-unpaired), без нуклеотидов с качеством чтения менее 20 в конце (TRAILING:20) и с минимальной длиной 50 (MINLEN:50).
Проверка качества триммированных чтений
Проверка осуществлялась с помощью программы fastqc, команды:
fastqc op1 fastqc ou1 fastqc op2 fastqc ou2
Количество участков секвенирования на парных выходах равно 39853177, что составляет приверно 96.27 процентов от изначального числа. У первого непарного - 678011 (1.64 процента), у второго - 659625 (1.59 процента), эта информация содержится в выводе trimmimatic из предыдущего пункта.
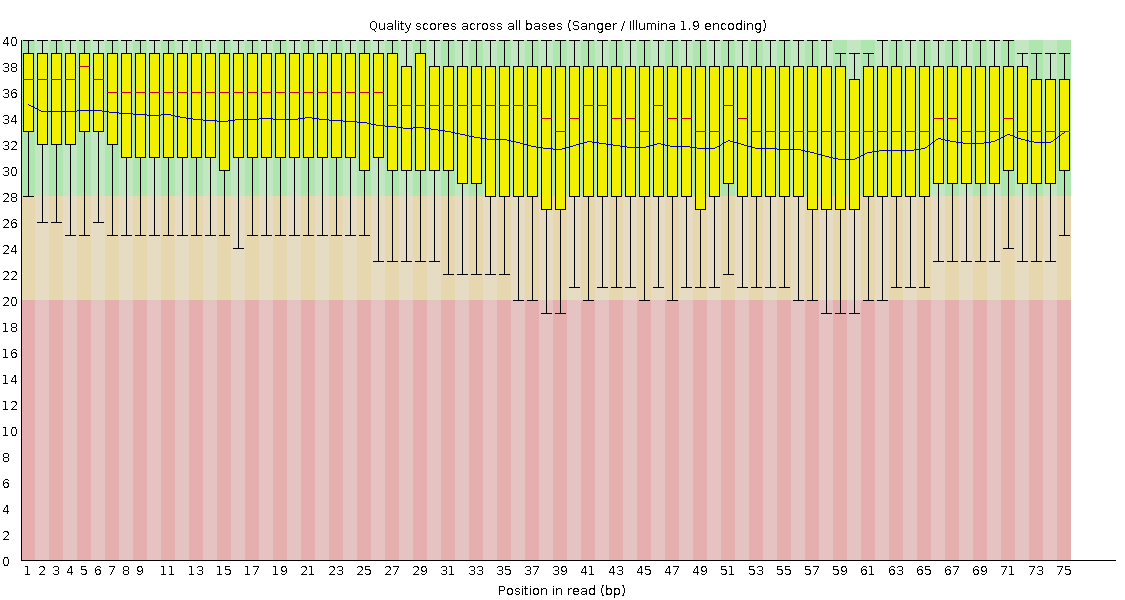
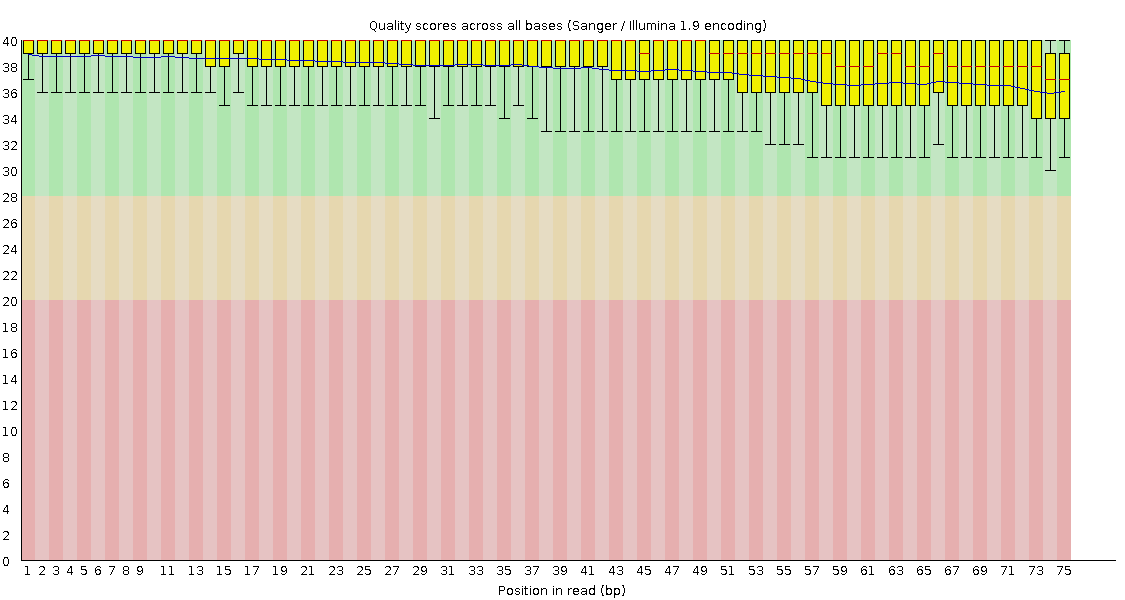
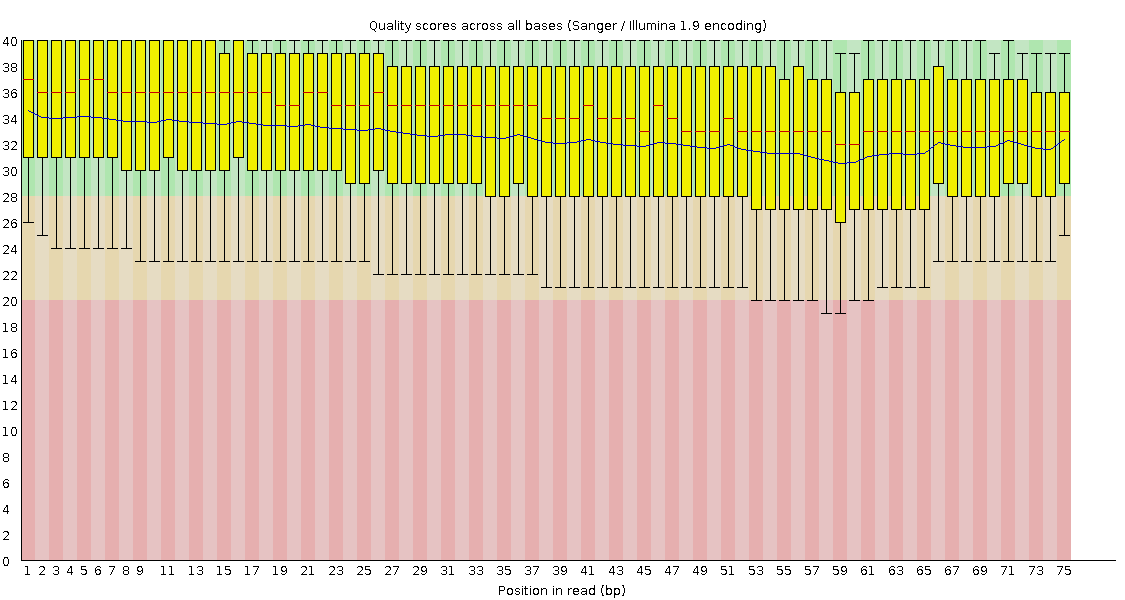
У "непарных" чтений (8, 10 рисунки) качество на 3-4 единицы ниже, чем у соответственных "парных" чтений. Возможно, "парные" чтения дают больше информации, и следовательно, более точные результаты.
По сравнению с необработанными trimmomatic данными, качество "парных" чтений немного на примерно на 1 единицу улучшилось (отклонение стало равномернее, около 4 единиц вниз для всех циклов элонгации), а про ухудшение качества "непарных" уже заявлено на абзац выше.
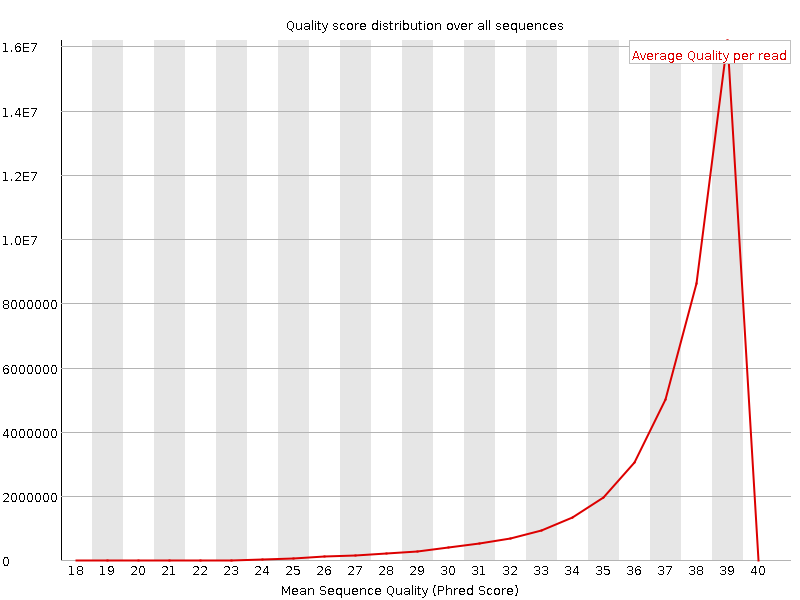
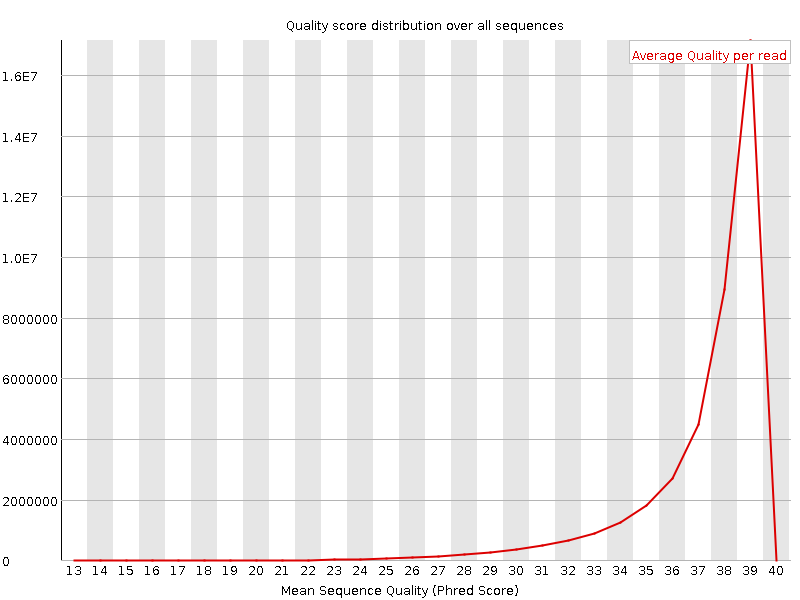
Распределение нуклеотидов с по качеству изменилось в основном за счет исчезновения из графика нуклеотидов с низким качеством (менее 17 в первом чтении и 13 во втором, при этом ожидалось, что нуклеотидов с качеством менее 20 не будет).
После использования trimmomatic появились чтения длиной менее 75, но из-за условия выполнения не менее 50 (на графике имеются небольшие количества с длиной 49). В остальном он не изменился.